

Portal Weekly #61: drugging the "undrugable", cold start problems in drug discovery, interactive molecular discovery, and more.
Portal Weekly #61: drugging the "undrugable", cold start problems in drug discovery, interactive molecular discovery, and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬




Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
⭐ Spotlight: Sign up for Polaris Updates
Our friends at Polaris has been on a roll the past few months!
Stay up to date on new releases on Polaris coming soon, including a potential upcoming hackathon by signing up for their mailing list 👀
From the launch event, to releasing Aurosis - a library that simplifies the curation process for drug discovery datasets, and Polaris Recipes - a resource promoting transparency and reproducibility, they have even more exciting initiatives to come.
Sign up here.

💬 Upcoming talks
Keep your eyes peeled on our LoGG calendar for upcoming talks! Join us live weekly in Zoom on Mondays at 11 am ET. Find more details here.
Speakers for CARE talks are usually announced on Fridays after this newsletter goes out. Sign up for Portal and get notifications when a new talk is announced on the CARE events page. You can also follow us on Twitter and LinkedIn!
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
ChatMol: Interactive Molecular Discovery with Natural Language
In biochemistry, tasks like predicting molecular properties and molecule mining are crucial but can be technically difficult. By linking natural language with chemical language, we can make these tasks more interpretable and accessible. This paper introduces conversational molecular design, which involves using natural language to describe and modify target molecules. To achieve this, the team developed ChatMol, a versatile and knowledgeable generative pre-trained model. ChatMol is enhanced by incorporating data on experimental properties, molecular spatial knowledge, and the relationships between natural and chemical languages.

DTI-LM: Language Model Powered Drug-Target Interaction Prediction
Ever heard of the cold start problem? In tech, it’s when a new product needs users for it’s worth to be defined. Turns out it’s an issue seen in predicting drug-target interactions (DTIs) as well. Using sequence-based representations of drugs and proteins in computational models has become popular due to their easy availability, simpler quality control, and lower computational demands. However, predicting DTIs for previously unknown drugs or proteins remains a tough challenge, especially for sequence-based models. This is where DTI-LM, a new framework, comes in. It utilizes advanced pre-trained language models to take advantage of their strong context-capturing abilities, combined with neighborhood information, to predict DTIs. In large-scale tests across four datasets, DTI-LM demonstrated top-tier performance in predicting DTIs. However, it was found that language models handle drug and protein similarities differently.

ML for Small Molecules
Computational design of serine hydrolases
Enzymes that carry out multistep reactions often rely on highly complex, precisely positioned polar active sites to perform specific chemical steps, making it incredibly difficult to design them from scratch. To tackle this challenge, researchers used the classic catalytic triad and oxyanion hole found in serine hydrolases as a model system. They employed RFdiffusion1 to create proteins with catalytic sites of increasing complexity and different geometries, while a new method called ChemNet was used to evaluate the geometry and preorganization of the active sites at each step of the reaction. Experimental testing revealed novel serine hydrolases that were able to catalyze ester hydrolysis with catalytic efficiencies (kcat/Km) up to 3.8 × 10³ M⁻¹ s⁻¹. These enzymes closely matched the design models, with Cα RMSDs under 1 Å, and featured protein folds that were distinct from those of natural serine hydrolases. By using in silico methods to select designs based on active site preorganization throughout the reaction process, the team significantly boosted success rates, identifying effective catalysts in screens of as few as 20 designs.

ML for Atomistic Simulations
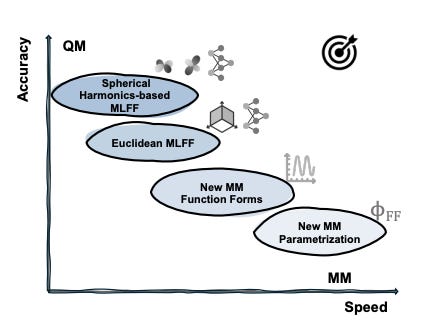
On the design space between molecular mechanics and machine learning force fields
What does quantum mechanics (QM) and molecular mechanics (MM) have in common with F1 drivers? The need for speed and accuracy. The ability to simulate a biomolecular system efficiently enough and meaningfully enough to get quantitative insights sounds like a dream, and it still is to this day. But machine learning force fields (MLFFs) represent a meaningful endeavour towards this direction, where differentiable neural functions are parametrized to fit ab initio energies, and furthermore forces through automatic differentiation. This review focuses on the design space (the speed-accuracy tradeoff) between MM and ML force fields. They dive into the desired properties and challenges now faced by the force field development community, survey the efforts to make MM force fields more accurate and ML force fields faster, and envision what the next generation of MLFF might look like.
Highly Accurate Real-space Electron Densities with Neural Networks
Variational ab-initio methods in quantum chemistry are unique in offering direct access to the wave function, enabling the extraction of any observable of interest, not just energy. However, in practice, extracting these observables can be technically challenging and computationally demanding. This paper focuses on electron density, a key observable in quantum chemistry, and presents a new method for obtaining accurate densities from real-space many-electron wave functions. By employing variational quantum Monte Carlo with deep-learning ansätze (deep QMC), they generate highly precise wave functions without basis set errors. They validated this approach by computing dipole moments, nuclear forces, contact densities, and other properties that rely on electron density.
ML for Proteins
Bridging the Gap between Sequence and Structure Classifications of Proteins with AlphaFold Models
Classifying protein domains based on homology and structural similarity is essential for understanding protein function. With recent breakthroughs in protein structure prediction, particularly through AlphaFold, the availability of structural data has dramatically increased. In this study, researchers aimed to classify around 9,000 Pfam families into the ECOD (Evolutionary Classification of Domains) system using predicted AlphaFold models and the DPAM (Domain Parser for AlphaFold Models) tool. More than half of the Pfam families contained DPAM domains that could be confidently classified within the ECOD hierarchy. By examining features such as disulfide bond patterns, metal-binding sites, and enzyme active sites, the team uncovered potential new structural folds and evolutionary relationships.

Computational Methods to Investigate Intrinsically Disordered Proteins and their Complexes
Wright and Dyson are not only a scientific power couple, but also brought to light that large sections of the proteome of all organisms are comprised of protein sequences that lack globular folded structures back in 1999. Since then, the biophysics community made strides in understanding intrinsically disordered proteins (IDPs) and intrinsically disordered regions (IDRs). This paper roundups an overview of current computational tools for IDPs and IDRs, and most recently their complexes and phase-separated states, including statistical models, physics-based approaches, and machine learning methods that permit structural ensemble generation and validation against many solution experimental data types.

ML for Omics
PROTAC-DB 3.0: an updated database of PROTACs with extended pharmacokinetic parameters
Proteolysis-targeting chimera (PROTAC) is a promising new therapeutic approach that uses the ubiquitin-proteasome system to break down proteins. Because of its unique, event-driven mechanism, PROTAC has the potential to target proteins that were previously considered "undruggable." Recently, the use of AI in drug design has sped up the development of PROTAC-based drugs. This paper presents an updated version of PROTAC-DB to version 3.0. This latest version has significantly expanded the database to include 6111 PROTACs, an 87% increase from version 2.0. It also features 569 warheads (small molecules that target the protein), 2753 linkers, and 107 E3 ligands (which help recruit E3 ligases). They have also added new sorting options based on molecular similarity and publication dates. You can access PROTAC-DB 3.0 here.

Reviews
Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋
 | A guest post by
|
 | A guest post by
|
 | A guest post by
|
 | A guest post by
|