

Portal Weekly #62: evaluating protein foundation models, single-cell data, making protein modeling accessible, and more.
Portal Weekly #62: evaluating protein foundation models, single-cell data, making protein modeling accessible, and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬
Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
💬 Upcoming talks
LoGG continues this week with a talk by Rob Brekelmans, a Postdoctoral Fellow at the Vector Institute in Toronto, to discuss Sequential Monte Carlo (SMC) for probabilistic inference problems. He’ll be able to answer all your questions about using SMC. Note that LoGG has changed from 11 am ET to 12 pm ET.

Join us live on Zoom on Monday at 12 pm ET. Find more details here.
Speakers for CARE talks are usually announced on Fridays after this newsletter goes out. Sign up for Portal and get notifications when a new talk is announced on the CARE events page. You can also follow us on Twitter and LinkedIn!
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
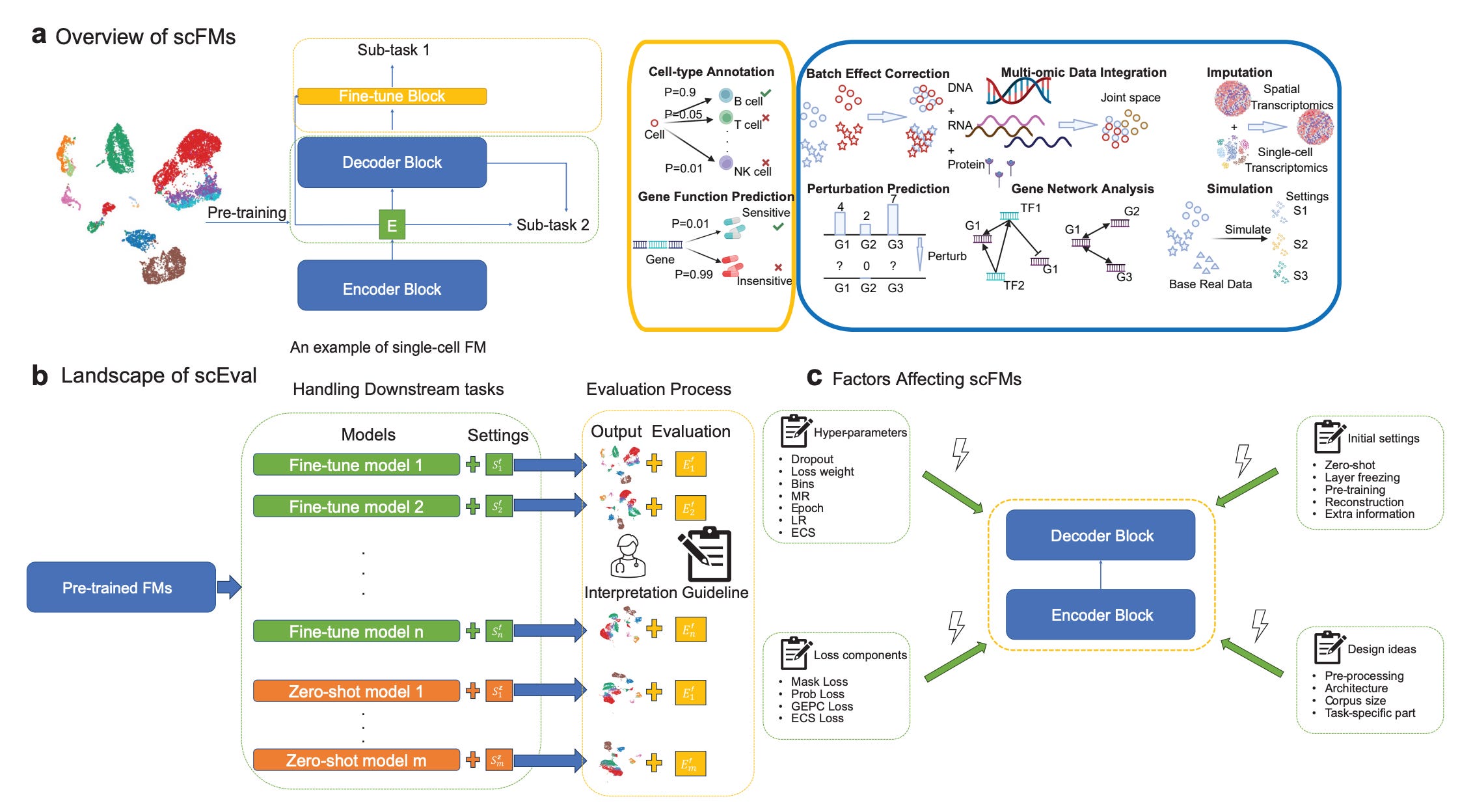
Evaluating the Utilities of Large Language Models in Single-Cell Data Analysis
Foundation Models (FMs) have made huge progress in industrial and scientific fields. This paper looks at the effectiveness of FMs for single-cell sequencing analysis through thorough experiments on eight downstream tasks relevant to single-cell data. The top-performing models, based on performance and ease of use, are scGPT, Geneformer, and CellPLM. They studied the impact of hyper-parameters, initial settings, and stability using a new framework called scEval and provided recommendations for improving performance through pre-training and fine-tuning.
Large Language Models in Drug Discovery and Development: From Disease Mechanisms to Clinical Trials
The use of Large Language Models (LLMs) in drug discovery and development is bringing about a major transformation. These models offer new approaches to understanding diseases, finding potential drugs, and improving clinical trials. This review discusses the growing importance of LLMs in various stages of the drug development process. It shows how they can uncover links between diseases and drug targets, interpret complex biomedical data, enhance drug design, predict drug safety and effectiveness, and make clinical trials more efficient. The paper is geared towards researchers in fields like computational biology, pharmacology, and AI, and provides insights into how LLMs could reshape drug discovery and development.
ML for Small Molecules
Lessons learned during the journey of data: from experiment to model for predicting kinase affinity, selectivity, polypharmacology, and resistance
Structure-based ML methods use models inspired by physics to predict how well a drug (ligand) will bind to its target protein. In this study, they talk about building KinoML, a new ML framework for discovering small molecule drugs that targets specific proteins, with a focus on structure-based methods. Right now, KinoML is focused on kinases, a large protein family, because their structures—especially in the kinase domain—are relatively conserved. KinoML lets users do three key things: access and curate molecular data, create features from that data that are suitable for ML, and run ML experiments that use information about ligands, proteins, and assays to predict how well a ligand will bind. Although KinoML currently focuses on kinases, the framework can be adapted to other proteins, and the lessons learned here could help guide the development of ML platforms in other areas of drug discovery.
Establishing the foundations for a data-centric AI approach for virtual drug screening through a systematic assessment of the properties of chemical data
In cheminformatics, researchers have been using advanced AI methods to handle the growing number of chemical libraries. Deep learning techniques have been shown to be more effective than traditional machine learning (ML) methods in tasks like QSAR and ligand-based virtual screening. However, these approaches often lack transparency and are hard to explain. Instead of focusing on more complex AI models (a model-centric approach), this team decided to explore the potential of a data-centric AI approach for virtual screening. They developed a new benchmark dataset of BRAF ligands and demonstrated that a conventional ML algorithm (SVM) with the right molecular representation (a combination of Extended and ECFP6 fingerprints) could achieve an incredible 99% accuracy in virtual screening. This far outperforms the results from more sophisticated deep learning methods. They also explored how the size and composition of the training data affect performance. They found that when inactive compounds outnumbered active ones, the model's recall decreased but precision increased, which lowered overall accuracy.
ML for Proteins
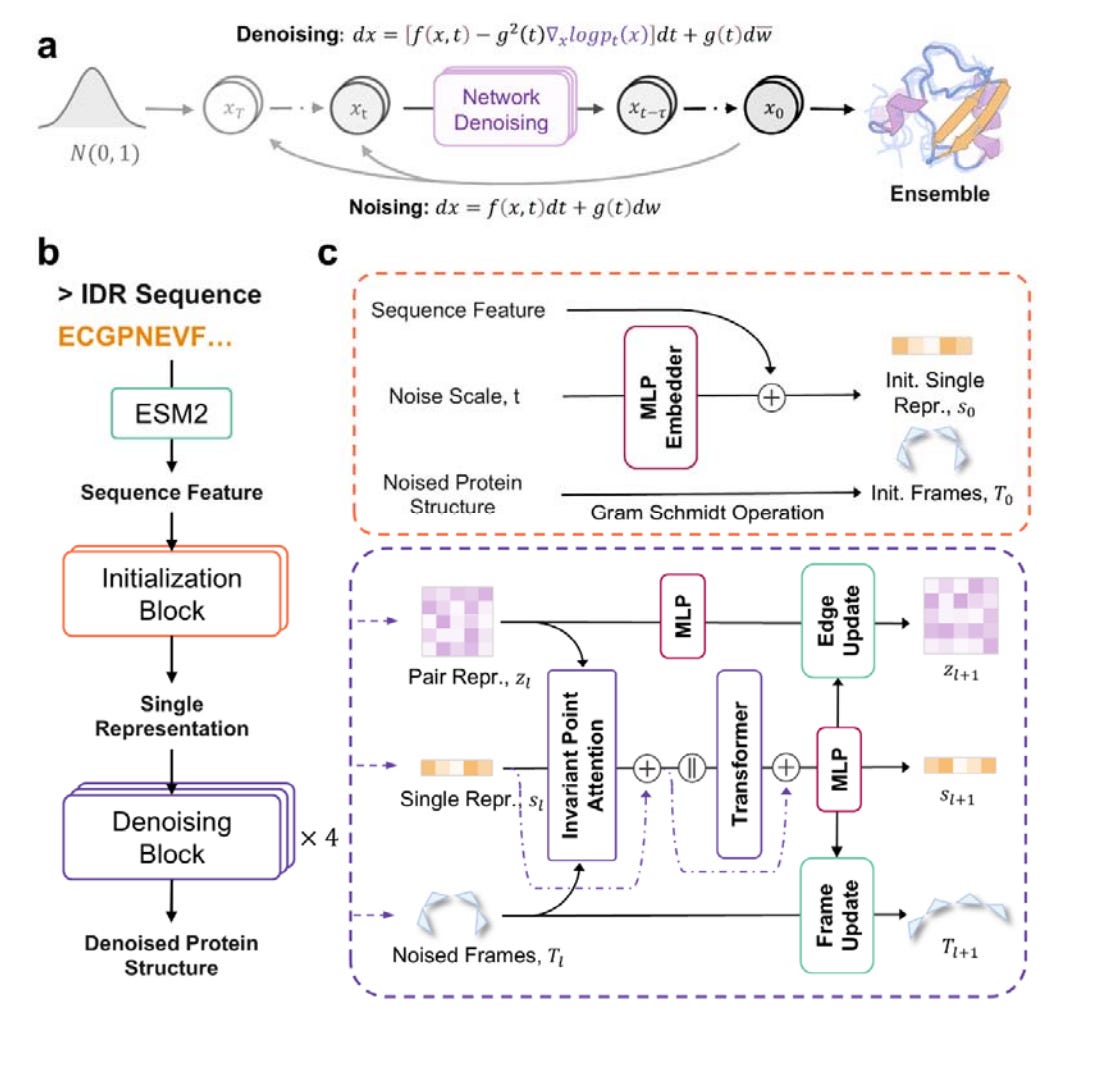
Precise Generation of Conformational Ensembles for Intrinsically Disordered Proteins via Fine-tuned Diffusion Models
Intrinsically disordered proteins, or IDPs, play a big role in various biological functions. Studying their structure usually involves a mix of molecular dynamics (MD) simulations and experimental data to help correct biases in the simulations. But the problem is that MD simulations are really expensive in terms of computing power, and there’s not enough experimental data to go around, which limits how much they can be used. That’s where IDPFold comes in. It’s a new tool that can generate different conformations for IDPs just from their sequences, using fine-tuned diffusion models. What’s cool about IDPFold is that it doesn’t need Multiple Sequence Alignments (MSA) or experimental data to work. Despite that, it still gives highly accurate predictions for a wide variety of IDPs. It goes a step further than other methods by focusing on the backbone structure, which leads to more detailed insights and better property predictions.
Enriching productive mutational paths accelerates enzyme evolution
Scientists are speeding up the evolution of enzymes, which are crucial for life, by mimicking natural selection. This paper used cycles of mutation, selection, and amplification, but instead of screening huge libraries of proteins, they focused on removing destabilizing mutations early on. This allowed them to evolve a specially designed enzyme (Kemp eliminase) that speeds up a key chemical reaction more than 108 times faster than without the enzyme. By combining this new variant with another evolved version, they found multiple ways to solve the same catalytic problem using different protein structures.

SaprotHub: Making Protein Modeling Accessible to All Biologists
Training and using deep learning models can be tough for people who aren't familiar with machine learning. SaprotHub is a platform that makes this whole process a lot easier. It allows biologists, even those without any ML experience, to train, use, and share protein ML models with just a few clicks. The platform encourages collaboration within the biology community. At the heart of it is Saprot, a powerful protein language model, which works through a tool called ColabSaprot. This setup supports hundreds of protein-related training and prediction tasks, making it easy for users to build, share, and improve models together, fostering more community-driven innovation

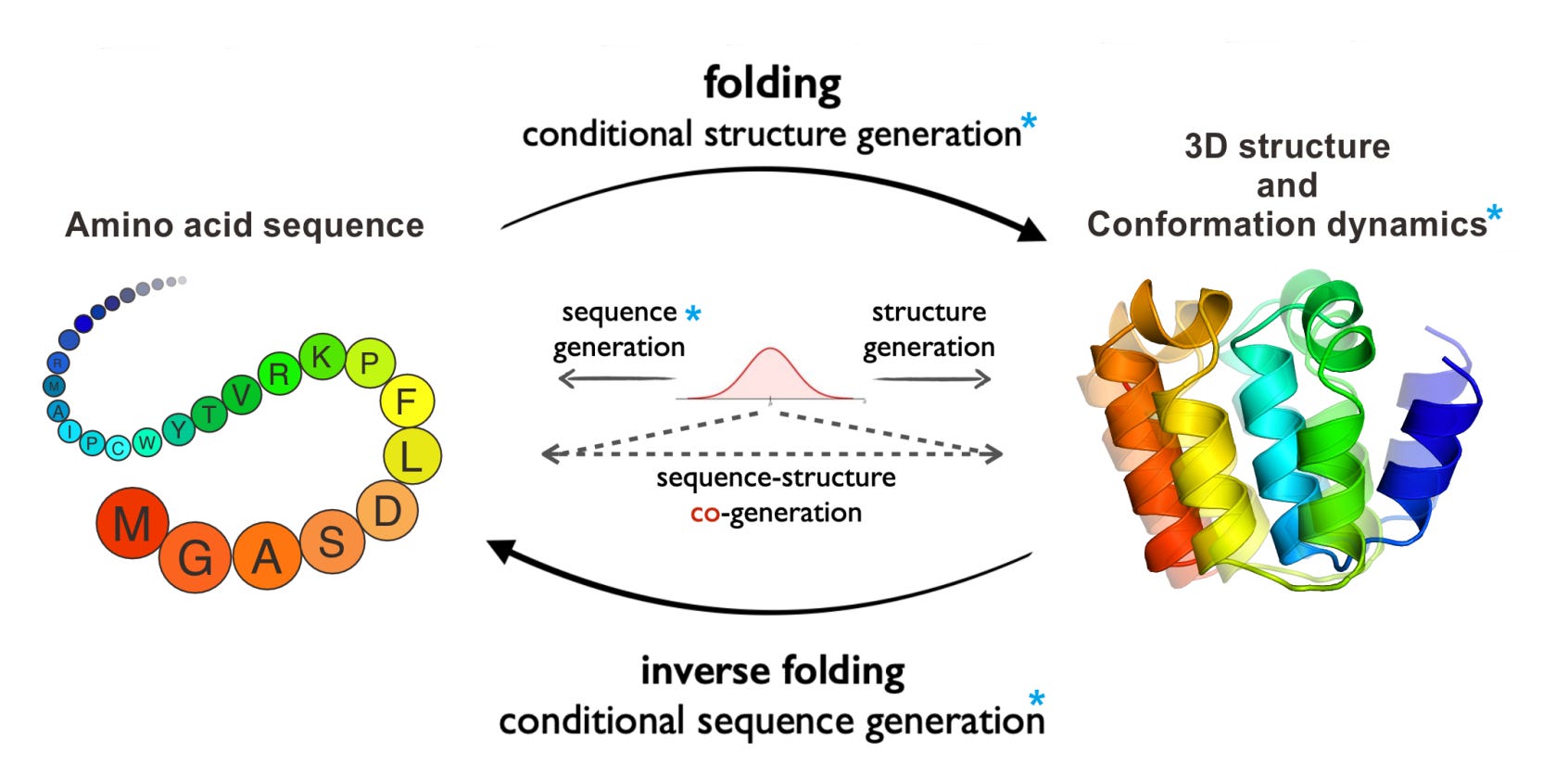
ProteinBench: A Holistic Evaluation of Protein Foundation Models
In recent years, there has been a rapid development of protein foundation models, greatly enhancing tasks like 3D structure prediction, protein design, and studying conformational dynamics. However, it's still unclear how well these models truly perform because there's no standard way to evaluate them. To address this, the researchers created ProteinBench, a comprehensive evaluation framework aimed at making protein foundation models more transparent. ProteinBench includes three main components: (i) a classification system for tasks that covers the key challenges in the protein field, based on the relationships between different protein types, (ii) a multi-metric evaluation system that measures performance in terms of quality, novelty, diversity, and robustness, and (iii) in-depth analyses that give a complete picture of model performance from different user perspectives. Their evaluation of protein models using ProteinBench uncovered important insights into what these models can and can’t do. To encourage more transparency and research, they’ve made the evaluation dataset, code, and a public leaderboard available here.
ML for Omics
A graph self-supervised residual learning framework for domain identification and data integration of spatial transcriptomics
Spatial transcriptomics (ST) technologies allow scientists to study gene expression within the context of tissue structure, but it’s still difficult to identify spatial patterns that match both gene expression and tissue histology, especially when working with data from multiple samples. To tackle this, the researchers developed ResST, a new model that uses graph neural networks and Margin Disparity Discrepancy (MDD) theory. ResST combines information about gene expression, biological factors, spatial location, and tissue structure to identify complex relationships between cells and their surroundings. What’s particularly useful about ResST is that it can work with multiple ST datasets and align them by correcting for batch effects using MDD theory. When tested on ten ST datasets from different technologies, ResST was able to identify spatial domains at a much finer scale. It also successfully integrated data from different tissue sections, whether stacked vertically or placed side-by-side, while correcting for inconsistencies between batches.

Reviews
Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
LoGG
Geometric deep learning framework for de novo genome by Lovro Vrček
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋