

Portal Weekly #63: biologically-inspired models for smell, virtual cells, ESMFold-predicted structures, and more.
Portal Weekly #63: biologically-inspired models for smell, virtual cells, ESMFold-predicted structures, and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬



Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
💬 Upcoming talks
LoGG continues next week with a talk by Carles Domingo-Enrich, Senior Researcher at Microsoft Research New England, to discuss stochastic optimal control (SOC) and a new algorithm named Adjoint Matching which outperforms existing SOC algorithms. He’ll be able to answer all your questions about this approach. Note that LoGG has changed from 11 am ET to 12 pm ET.

Join us live on Zoom on Monday at 12 pm ET. Find more details here.
Speakers for CARE talks are usually announced on Fridays after this newsletter goes out. Sign up for Portal and get notifications when a new talk is announced on the CARE events page. You can also follow us on Twitter and LinkedIn!
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
How to Build the Virtual Cell with Artificial Intelligence: Priorities and Opportunities
The cell is the smallest unit of life, and if we really want to understand biology, we have to understand cells. That’s why it’s so important to be able to accurately model them—not just for understanding how they work, but also to figure out the root causes of diseases. In this paper, the authors lay out a vision for what they call AI-powered "Virtual Cells." The idea is that AI could help us create detailed, reliable representations of cells and their systems under all kinds of conditions, using the massive amounts of biological data being generated. They talk about how these AI-driven Virtual Cells could give us a universal way to represent biological processes across different scales, from molecules to whole systems, and also make it easier to run virtual experiments that would help predict how cells behave. The ultimate goal? A future where AI-powered Virtual Cells can help us find new drug targets, predict how cells will react to changes, and explore scientific hypotheses on a much bigger scale.

ML for Small Molecules
MolPipeline: A Python package for processing molecules with RDKit in scikit-learn
MolPipeline is a new Python package that brings together RDKit and scikit-learn to make molecular machine learning much easier. It automates the entire workflow, from processing molecular data all the way to creating models that are ready to use. One of the big advantages of MolPipeline is how it handles large datasets—it can process them in parallel with minimal memory usage, which is great for dealing with big data. It’s also super flexible, letting users build custom pipelines for various tasks in cheminformatics, like cleaning up molecules, extracting features, and clustering them. A standout feature is its error handling. If a molecule is unreadable, like a messed-up SMILES string, it’ll automatically catch the error, log it, and replace it with a placeholder value. MolPipeline also comes with built-in tools for common cheminformatics tasks, like extracting Murcko scaffolds, cleaning up molecules (removing salts or handling tautomers), and converting molecules into different formats like SMILES or fingerprints. There’s comprehensive documentation and examples, so whether you're new to this field or an experienced user, MolPipeline is easy to pick up and run with.

ML for Atomistic Simulations
All-in-one foundational models learning across quantum chemical levels
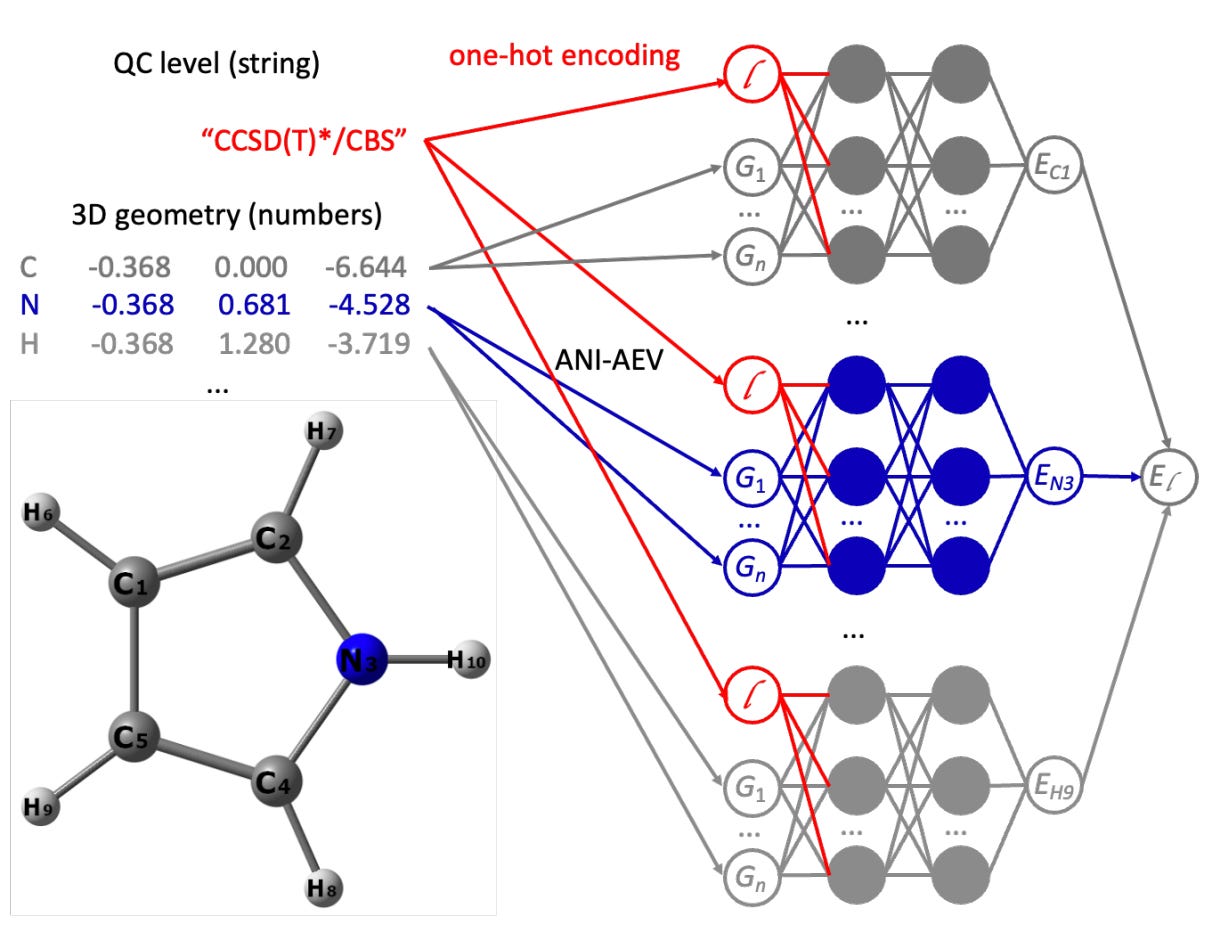
Machine learning (ML) models usually focus on just one quantum chemical (QC) level, and the models built for multi-fidelity learning haven't really scaled well for foundational models. That's where this paper’s all-in-one (AIO) ANI model architecture comes in. It uses multimodal learning to handle any number of QC levels, making it a simpler and more versatile alternative to transfer learning. They have used this approach to create the AIO-ANI-UIP foundational model, which has a generalization ability comparable to methods like GFN2-xTB and DFT with a double-zeta basis set, specifically for organic molecules. What's exciting is that the AIO-ANI model can learn across various QC levels, from semi-empirical methods to density functional theory and even coupled cluster.
ML for Proteins
Accurately predicting enzyme functions through geometric graph learning on ESMFold-predicted structures
GraphEC is a novel enzyme prediction tool that improves upon existing methods by focusing on enzyme active sites and structural features. It uses geometric graph learning with ESMFold-predicted structures and a pre-trained protein language model to predict Enzyme Commission (EC) numbers. The model first identifies active sites and then predicts EC numbers, refining these predictions with homology information through a label diffusion algorithm. Additionally, GraphEC predicts the optimum pH for enzyme-catalyzed reactions. Experiments show it outperforms current techniques in predicting active sites, EC numbers, and optimum pH, with potential applications in synthetic biology and genomics.

NeoaPred: A deep-learning framework for predicting immunogenic neoantigen based on surface and structural features of peptide-HLA complexes
NeoaPred brings a fresh approach to predicting immunogenic neoantigens by combining both surface and structural features of peptide-HLA (pHLA) complexes. This is a big step forward from traditional methods that only rely on sequence-based predictions. One standout feature is the use of PepConf, which predicts the structure of peptide-HLA-I complexes with an impressive 82.37% accuracy and an RMSD under 1Å. NeoaPred also has a PepFore module that calculates a 'foreignness score,' based on surface and structural differences between mutant and wild-type peptides. This score has proven to be a strong indicator of immunogenicity, outperforming other methods with an AUROC of 0.81. This model greatly improves neoantigen identification, which is critical for cancer immunotherapy. While it's highly effective across many peptide-HLA alleles, it’s less accurate for HLA-C alleles, mostly due to limited training data.

ML for Omics
Mapping the combinatorial coding between olfactory receptors and perception with deep learning
The sense of smell is still a bit of a mystery, especially when compared to how well we understand vision and hearing. At the core of how we smell are odorant molecules, which trigger specific olfactory receptors (ORs) in our nose. The unique combination of these activated receptors helps code for the specific smells we perceive. However, predicting how molecules interact with these receptors has proven to be extremely difficult. In this work, the author introduces a new, biologically-inspired approach that tackles this challenge. First, they map odorant molecules to their corresponding OR activation profiles, and then use these profiles to predict how the odorants will be perceived. They show that their model is able to distinguish binding patterns across different OR families, as well as between active protein-coding genes and frequently occurring pseudogenes in the human olfactory genome. This research could lead to the discovery of new odorant molecules that interact with orphan ORs (receptors whose functions are still unknown) and help deepen our understanding of the relationship between chemical structures and the smells they produce.

Protein evolution and data-driven sequence landscapes
Thanks to the explosion of available protein sequence data, largely driven by next-generation sequencing, unsupervised machine learning models have become powerful tools for understanding protein sequence landscapes. Specifically, Direct Coupling Analysis (DCA) is a method that directly examines patterns of conservation and coevolution between protein sites. In this thesis, the author extends the use of DCA to study protein evolution, which is the process by which amino acid substitutions occur in proteins due to random mutations and natural selection. The focus is on understanding epistasis, where the effects of one mutation depend on the presence of others. The author begins by modeling two recent neutral drift experiments, which used beta-lactamases as starting sequences. Next, the author investigates deep mutational scanning data from two other beta-lactamase enzymes, VIM-2 and NDM-1, and analyzes the context-dependent variability of mutations. Finally, they introduce an algorithm that models protein evolution across different timescales, providing a way to quantify how the predictability of mutational effects diminishes as sequences diverge, a phenomenon driven by epistasis.

Reviews
Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
LoGG
Probabilistic Inference in Language Models via Twisted Sequential Monte by Rob Brekelmans
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋
 | A guest post by
|
 | A guest post by
|
 | A guest post by
|