

Portal Weekly #64: bayesian optimization, assessing chemical tasks, full-atom time-coarsened dynamics, functional antibody design, and more.
Portal Weekly #64: bayesian optimization, assessing chemical tasks, full-atom time-coarsened dynamics, functional antibody design, and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬



Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
📅 BIOxML Hackathon Deadline Extended
Join Lux Capital, Evolutionary Scale, & Enveda Sciences in the second Bio x ML Hackathon to drive forward the frontier of science. Build on top of latest foundation models like ESM3 (98B params, GPU access) & proprietary datasets to develop next-gen biology applications.
Apply by Saturday Sep 28 11:59 pm PT. Check out more details here.

💬 Upcoming talks
Make sure to check the LoGG calendar for future talks updates. Join live on Zoom on Mondays at 12 pm ET. Find more details here.
You can also follow us on Twitter and LinkedIn!
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
Large Language Models to Enhance Bayesian Optimization
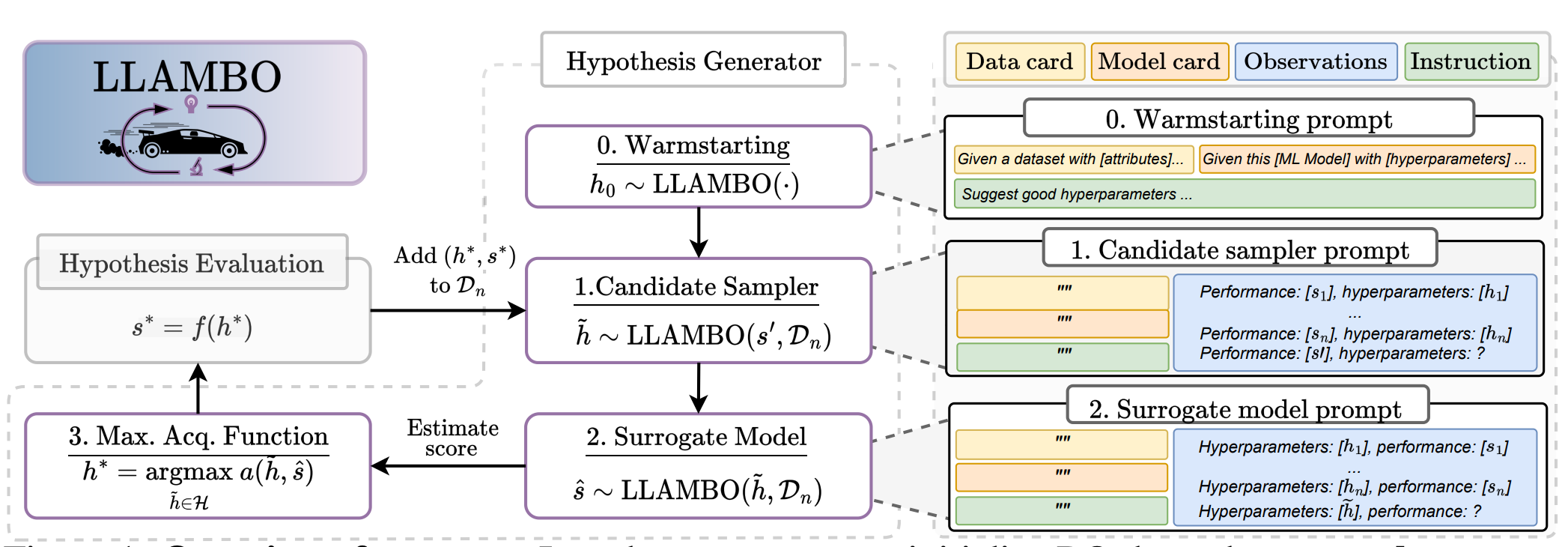
Bayesian optimization (BO) is a powerful method for optimizing complex and expensive-to-evaluate black-box functions, and it plays a crucial role in various applications like hyperparameter tuning. However, its effectiveness hinges on efficiently balancing exploration and exploitation, which remains a delicate challenge despite significant advancements in BO techniques. The authors introduce LLAMBO, a novel approach that incorporates the capabilities of Large Language Models (LLMs) into BO. Their findings demonstrate that LLAMBO is effective at zero-shot warm-starting and improves surrogate modeling and candidate sampling, especially during the early stages of search when data is sparse. Its modular design allows individual components to be integrated into existing BO frameworks or to function cohesively as a comprehensive, end-to-end solution.
ChemEval: A Comprehensive Multi-Level Chemical Evaluation for Large Language Models
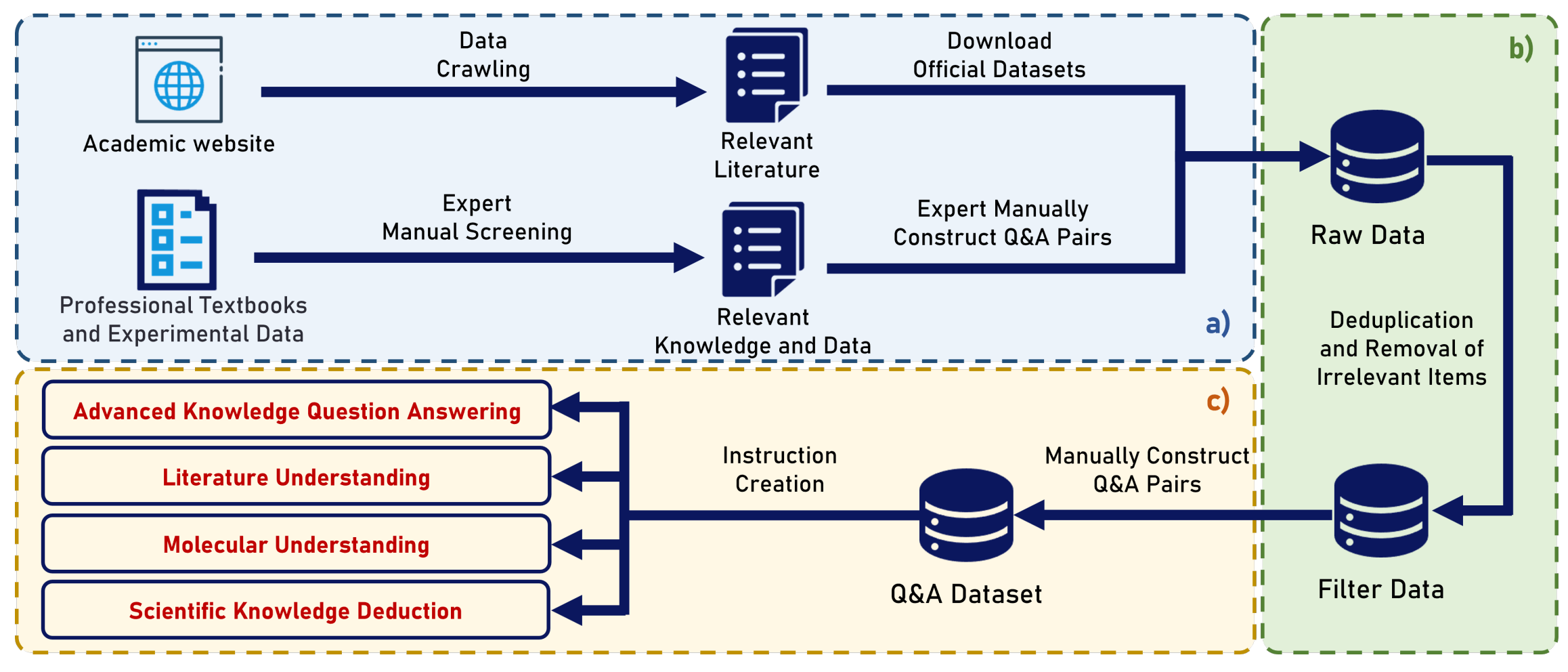
Introducing ChemEval, a comprehensive benchmark designed to assess the capabilities of LLMs across a wide range of chemical tasks. ChemEval encompasses four progressive levels in chemistry, evaluates 12 dimensions of LLM performance, and includes 42 distinct tasks informed by open-source data and datasets crafted by chemical experts to ensure practical relevance. In their experiments, the authors evaluated 12 mainstream LLMs using ChemEval under zero-shot and few-shot learning conditions, employing carefully selected examples and prompts. The results indicate that general LLMs like GPT-4 and Claude-3.5 perform well in literature understanding and following instructions but struggle with tasks requiring advanced chemical knowledge. Conversely, specialized LLMs demonstrate stronger chemical competencies but have reduced proficiency in literary comprehension.
ML for Atomistic Simulations
Force-Guided Bridge Matching for Full-Atom Time-Coarsened Dynamics of Peptides
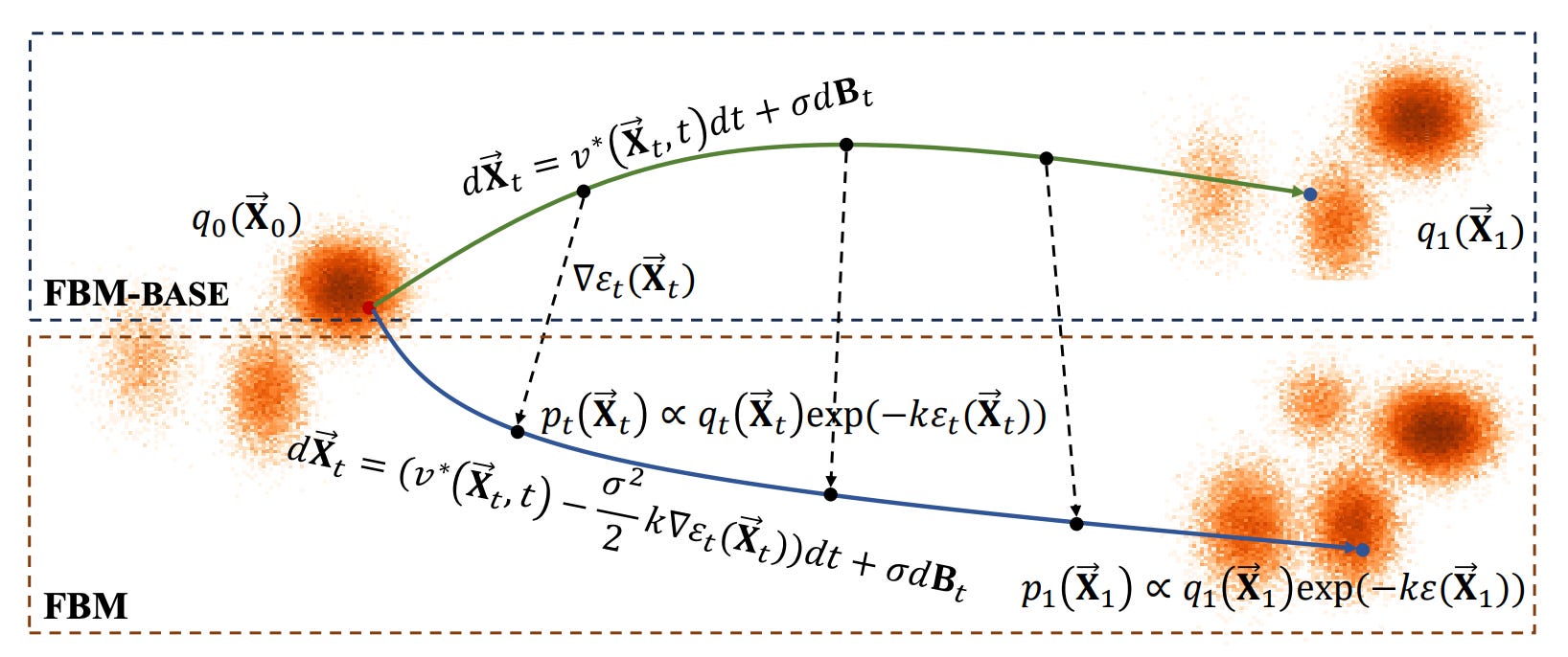
The authors address the challenges in Molecular Dynamics (MD) simulations, which are essential in fields like materials science, chemistry, and pharmacology but often struggle with balancing time cost and prediction accuracy. Traditional MD software faces limitations that hinder wider application due to these trade-offs. To improve efficiency and universality over longer time steps, recent data-driven approaches have utilized deep generative models for time-coarsened dynamics. To overcome these limitations, the authors propose a conditional generative model called Force-guided Bridge Matching (FBM). FBM learns full-atom time-coarsened dynamics while targeting the Boltzmann-constrained distribution. It incorporates a carefully designed intermediate force field to guide the generation process, effectively leveraging physics priors to enhance simulations.
ML for Small Molecules
Modeling protein-small molecule conformational ensembles with ChemNet

The authors tackle the challenge of modeling conformational heterogeneity in protein-small molecule systems by introducing ChemNet, a graph neural network that operates at the atomic level. Trained on structures from the Cambridge Structural Database and the Protein Data Bank, ChemNet accurately reconstructs atomic positions with atoms as graph nodes. This allows it to generate structures of diverse organic small molecules and protein side chains for docking purposes. Its rapid and stochastic nature enables the creation of prediction ensembles to map conformational variability. In enzyme design efforts, using ChemNet to evaluate the accuracy and pre-organization of designed active sites led to higher success rates and increased activities; notably, they achieved a preorganized retroaldolase with a kcat/K_M of 11,000 M⁻¹·min⁻¹, surpassing previous designs without deep learning.
PromptSMILES: prompting for scaffold decoration and fragment linking in chemical language models

This paper presents a simple procedure to extend the applicability of SMILES-based generative models in drug design to tasks like scaffold decoration and fragment linking without the need for retraining new models. While these models are typically used for complete de novo molecule generation, adapting them to scaffold decoration and fragment linking usually requires different grammars, architectures, and training datasets. By providing SMILES prompts and combining them with reinforcement learning, they show that pre-trained, decoder-only models can quickly adapt to these applications and optimize molecule generation toward specified objectives. Their approach performs comparably or better than a variety of other methods, and they offer an easy-to-use Python package to facilitate model sampling, available on GitHub and the Python Package Index.
ML for Proteins
Modeling Boltzmann weighted structural ensembles of proteins using AI based methods

The authors of this review highlight recent advances in AI-driven methods for generating Boltzmann-weighted structural ensembles, which are crucial for understanding biomolecular dynamics and facilitating drug discovery. They discuss how the rise of deep learning models like AlphaFold2 has led to more accurate and efficient sampling of these ensembles. The review covers the integration of AI with traditional molecular dynamics techniques and experimental data, addresses the challenges of conformational sampling, and explores future directions for AI-driven research in structural biology, particularly in the areas of drug discovery and protein dynamics.
IgGM: A Generative Model for Functional Antibody and Nanobody Design

The authors introduce IgGM, a generative model that combines diffusion and consistency models to generate antibodies with functional specificity, addressing challenges in integrating AI-driven antibody design into real-world processes. Unlike existing methods that require unrealistic conditions, IgGM simultaneously produces antibody sequences and structures for a given antigen using three core components: a pre-trained language model for extracting sequence features, a feature learning module for identifying pertinent features, and a prediction module that outputs designed antibody sequences along with the predicted complete antibody-antigen complex structure. The model has demonstrated effectiveness in both predicting structures and designing novel antibodies and nanobodies, making it relevant for practical applications in antibody and nanobody design.
Zero-shot transfer of protein sequence likelihood models to thermostability prediction

The authors investigate protein sequence likelihood models (PSLMs), a new class of self-supervised deep learning algorithms that learn probability distributions over amino acid identities based on structural or evolutionary context. While PSLMs have recently shown impressive performance in predicting the relative fitness of variant sequences without task-specific training, their potential to enhance protein stability—a central goal in protein engineering—has been underexplored. In this study, they comprehensively analyze the zero-shot transfer capability of eight PSLMs for predicting relative thermostability across hundreds of protein variants using several quantitative datasets. Comparing PSLMs with popular task-specific stability models, they find that some PSLMs perform competitively when appropriate statistics are considered. They highlight the strengths and weaknesses of PSLMs, examine their complementarity with task-specific models, and focus on applications in stability engineering.
ML for Omics
PRAGA: Prototype-aware Graph Adaptive Aggregation for Spatial Multi-modal Omics Analysis
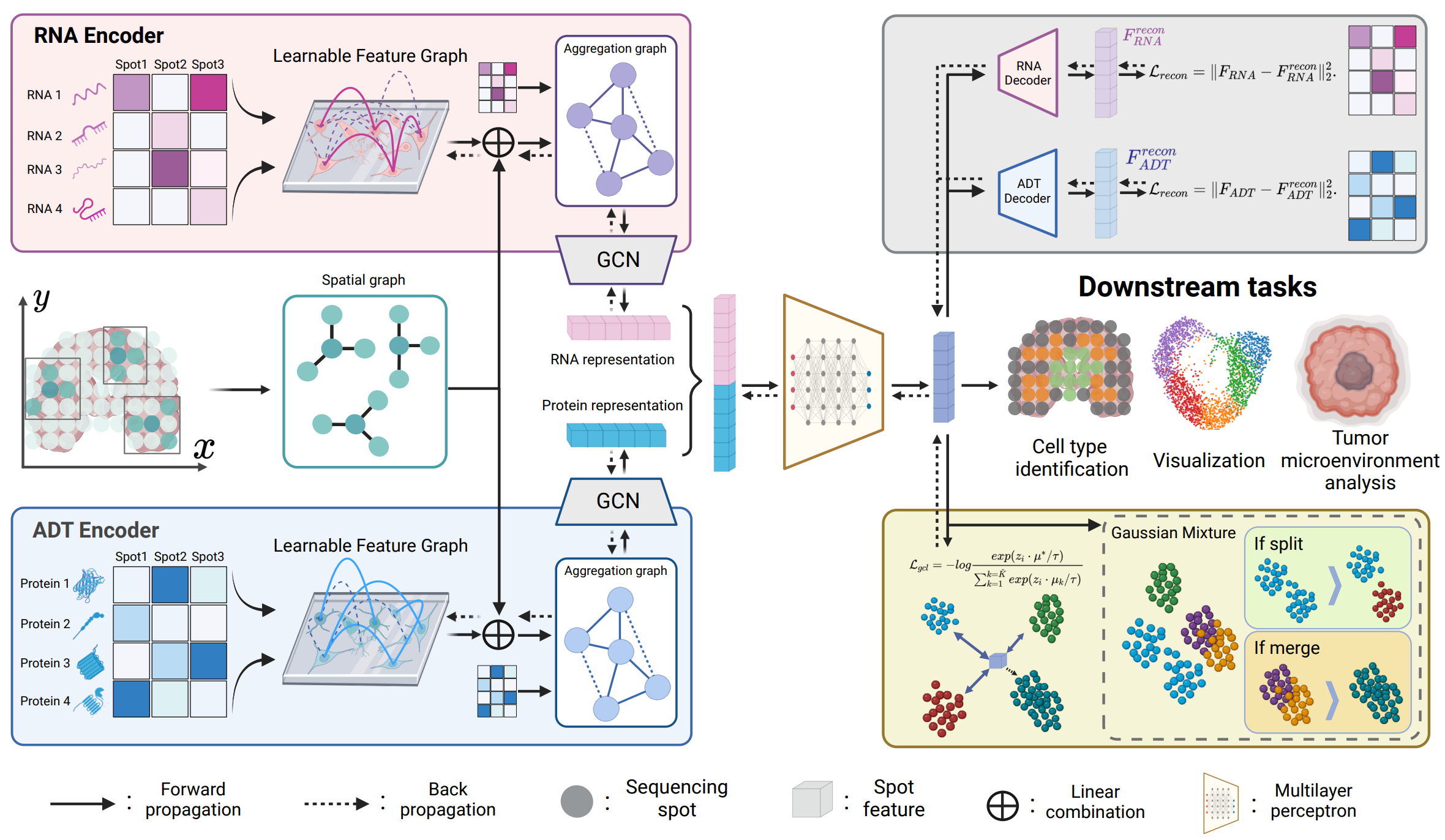
Did you know in Nature Methods 2023 spatial multi-modal omics technology was recognized as advanced biological technique essential for understanding biological regulatory processes within their spatial context. Recently, graph neural networks (GNNs) based on K-nearest neighbor (KNN) graphs have become prominent in spatial multi-modal omics methods due to their ability to model semantic relationships between sequencing spots. However, fixed KNN graphs fail to capture latent semantic relations hidden by data perturbations during the biological sequencing process, leading to a loss of semantic information. To address these challenges, the authors propose a novel framework called PRototype-Aware Graph Adaptive Aggregation for Spatial Multi-modal Omics Analysis (PRAGA). PRAGA constructs a dynamic graph to capture latent semantic relations and integrates spatial information with feature semantics comprehensively. Moreover, they introduce a dynamic prototype contrastive learning method based on the adaptability of Bayesian Gaussian Mixture Models to optimize multi-modal omics representations without relying on known biological priors.
Open Source
Open Source Infrastructure for Automatic Cell Segmentation
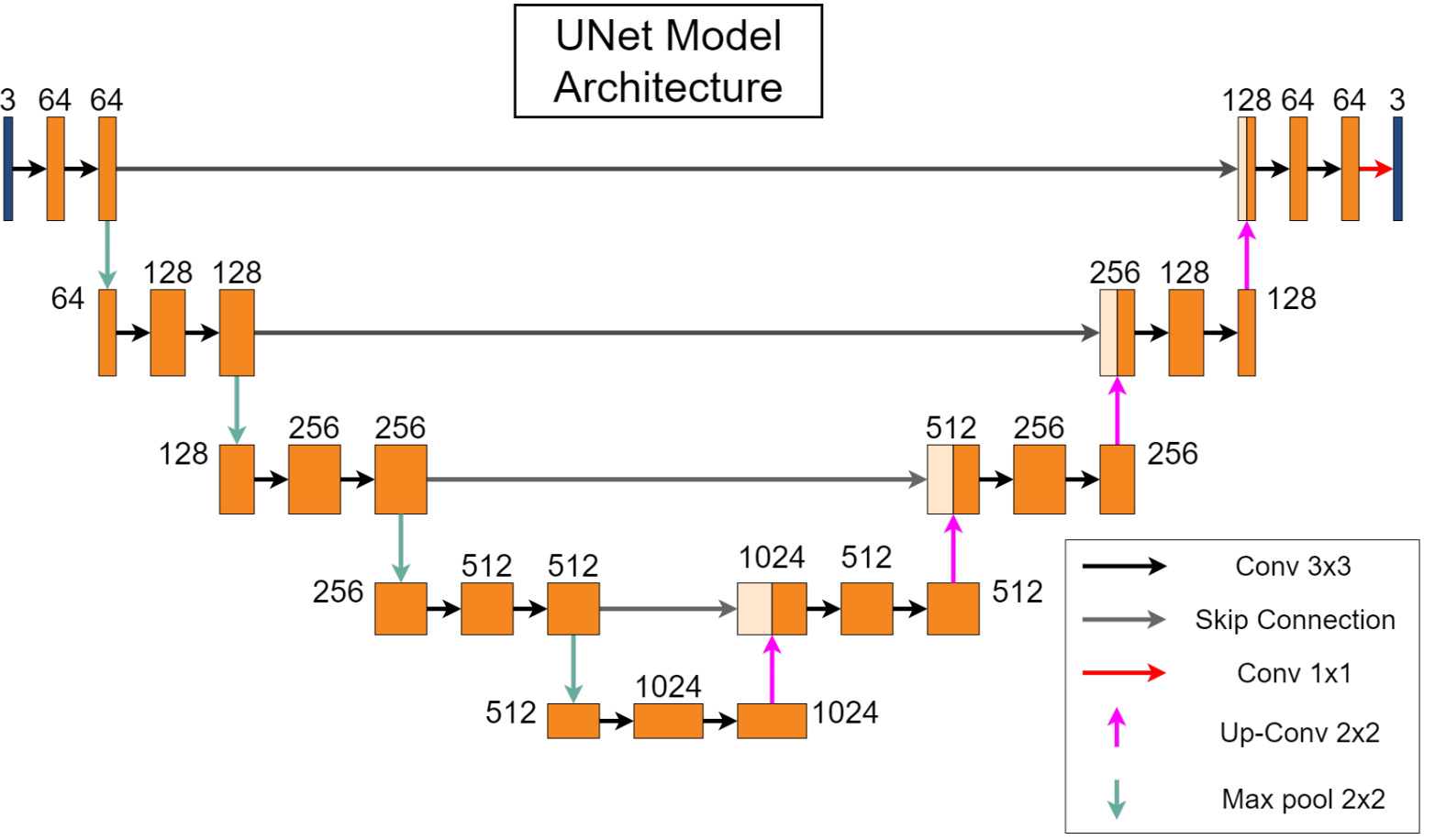
There is a growing importance of automated cell segmentation in various biological and medical applications, including cell counting, morphology analysis, and drug discovery. Manual segmentation is both time-consuming and subjective, underscoring the need for robust automated methods. In this paper, they present an open-source infrastructure that utilizes the UNet model, a deep-learning architecture known for its effectiveness in image segmentation tasks. This implementation is integrated into the open-source DeepChem package, enhancing accessibility and usability for researchers and practitioners. The resulting tool provides a convenient and user-friendly interface, lowering the barrier to entry for cell segmentation while maintaining high accuracy. Additionally, they benchmark this model against various datasets, demonstrating its robustness and versatility across different imaging conditions and cell types.
Reviews
Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
LoGG
Adjoint Matching: Fine-tuning Flow and Diffusion Generative Models with Memoryless Stochastic Optimal by Carles Domingo-Enrich
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋