


Portal Weekly #66: inverse molecular design, text-guided diffusion model, orbital inspired molecular representation, and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬



Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
💬 Upcoming talks
LoGG continues next week with a talk by Xiner Li and Masatoshi Uehara to discuss an iterative sampling method using soft value functions to predict rewards in diffusion models. They’ll be able to answer all your questions. Please note that LoGG has changed from 11 am ET to 12 pm ET.

Join us live on Zoom on Monday at 12 pm ET. Find more details here.
Speakers for CARE talks are usually announced on Fridays after this newsletter goes out. Sign up for Portal and get notifications when a new talk is announced on the CARE events page. You can also follow us on Twitter and LinkedIn!
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
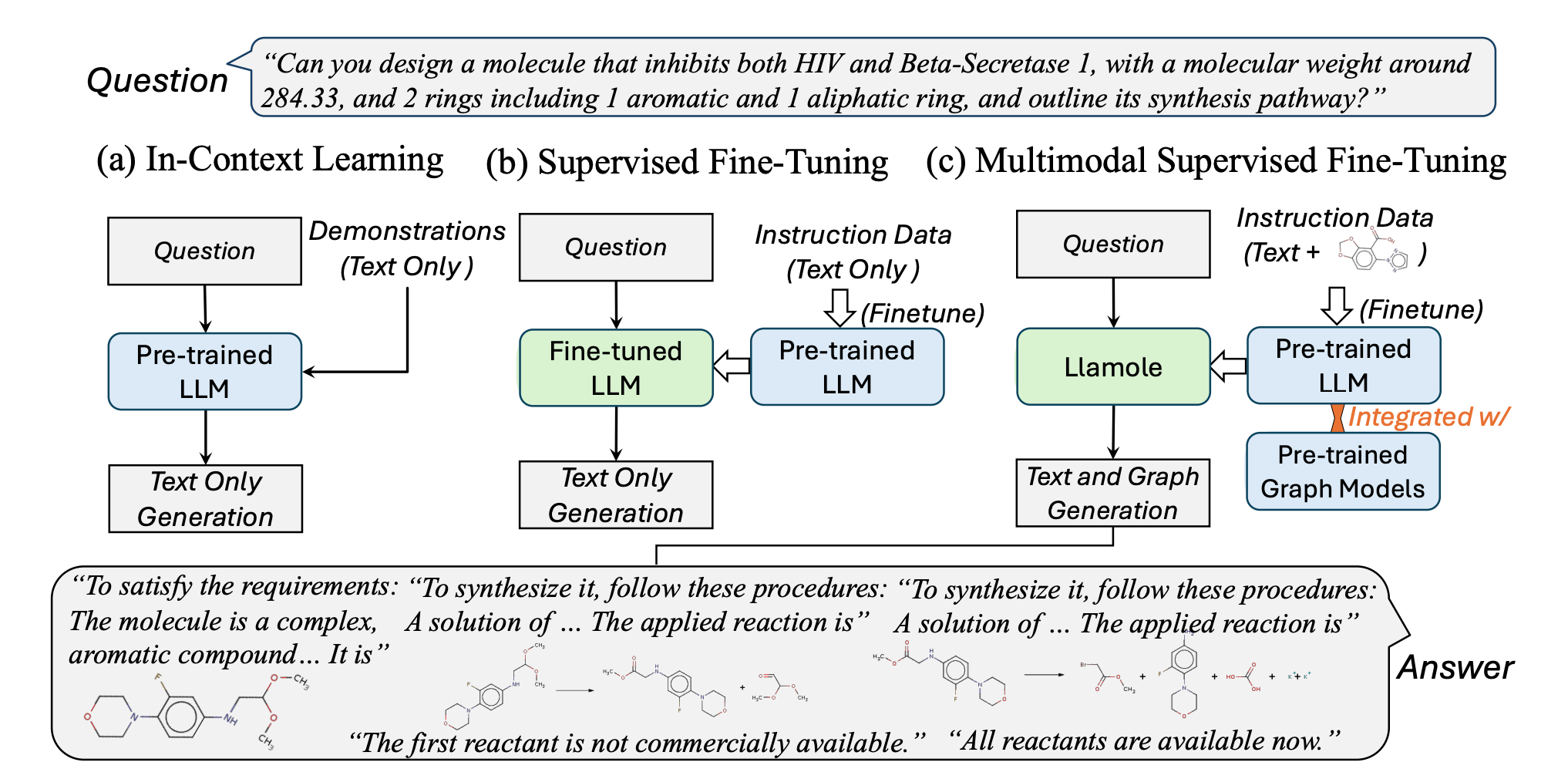
Multimodal Large Language Models for Inverse Molecular Design with Retrosynthetic Planning
Large language models (LLMs) have integrated images but struggle with adapting to graphs, limiting their use in areas like materials and drug design. To overcome this, the paper introduces Llamole (Large language model for molecular discovery) the first multimodal LLM that can generate both text and graphs, enabling molecular inverse design with retrosynthetic planning. Llamole combines a base LLM with Graph Diffusion Transformers and Graph Neural Networks for multi-conditional molecular generation and reaction inference. It also integrates A* search with LLM-based cost functions for efficient retrosynthetic planning. Through extensive experiments, Llamole outperformed 14 adapted LLMs across 12 metrics, proving superior in controllable molecular design and retrosynthetic planning. This advancement opens up new possibilities for combining text and graph generation in complex scientific applications.
Conditional Enzyme Generation Using Protein Language Models with Adapters
The conditional generation of proteins with specific functions or properties is a key goal in the development of generative models. Current methods, which use language models with simple prompts, can generate proteins based on a target functionality, like a specific enzyme family. However, these approaches are limited by basic, token-based conditioning and haven't been shown to work for unseen functions. In this study, researchers introduce ProCALM (Protein Conditionally Adapted Language Model), a new method for generating proteins conditionally, using adapters for protein language models. Specifically, they fine-tuned ProGen2 to incorporate conditioning based on enzyme function and taxonomy. ProCALM performs on par with existing methods in generating sequences from targeted enzyme families, but it goes a step further by generating sequences within the joint distribution of enzyme function and taxonomy. Impressively, it can also generalize to rare and previously unseen enzyme families and taxonomies.

ML for Small Molecules
Generative Flows on Synthetic Pathway for Drug Design
Generative models are transforming drug discovery, but many fail to account for synthesizability, limiting practical use. RXNFLOW addresses this by assembling molecules with predefined building blocks and chemical reaction templates, ensuring realistic synthetic pathways. Using Generative Flow Networks (GFlowNets) and a novel action space subsampling method, RXNFLOW efficiently explores vast molecular combinations, generating diverse and high-reward molecules without computational strain. It adapts to new objectives and building blocks without retraining, and outperforms existing models in pocket-specific optimization, achieving state-of-the-art results on CrossDocked2020, including a Vina score of –8.85 kcal/mol and 34.8% synthesizability. RXNFLOW offers a robust solution for practical, synthesizable drug design.

Text-guided Diffusion Model for 3D Molecule Generation
De novo molecule design is key to drug discovery, enabling the creation of molecules with specific structures for targeted properties. However, manually designing molecules within the vast chemical space is a significant challenge. Recent advances in machine learning, particularly diffusion models, have shown promise in automating this process, yet many generated molecules fail to meet practical needs like synthesizability or desired biological activity. To address this, the paper introduces TextSMOG, a novel Text-guided Small Molecule Generation Approach. TextSMOG integrates advanced language models with equivariant diffusion models (EDM) to enable the creation of 3D molecular structures based on natural language descriptions. TextSMOG was tested on datasets like QM9 and PubChem, where it outperformed existing diffusion-based models by generating molecules that are not only stable and diverse but also align with specific textual conditions. The model achieved higher scores in synthetic accessibility and Tanimoto similarity, proving its effectiveness in generating valid 3D molecular conformations.

ML for Atomistic Simulations
End-to-End Reaction Field Energy Modeling via Deep Learning based Voxel-to-voxel Transform
In computational biochemistry, understanding electrostatic interactions is key to analyzing biomolecules' structure, dynamics, and function. The Poisson-Boltzmann (PB) equation is crucial for modeling these interactions but is computationally demanding, especially for complex biomolecular surfaces and mobile ions. Traditional numerical methods, though accurate, scale poorly with larger systems. To address this, the paper introduces PBNeF, a machine learning-based solution inspired by neural network approaches for solving partial differential equations. PBNeF converts the PB equation's input and boundary conditions into a learnable voxel representation and uses a neural field transformer to predict the PB solution and reaction field potential energy. Experiments show PBNeF delivers over 100x speedup compared to traditional PB solvers, with accuracy on par with the Generalized Born (GB) model, making it a breakthrough in efficient electrostatic modeling.

E3STO: Orbital Inspired SE(3)-Equivariant Molecular Representation for Electron Density Prediction
Electron density prediction is crucial for understanding molecular interactions and conducting quantum mechanical calculations, but traditional Density Functional Theory (DFT) is computationally expensive. To address this, the paper introduces a novel SE(3)-equivariant architecture inspired by Slater-Type Orbitals (STO) to efficiently learn molecular electronic structures. This approach provides an alternative, orbital-like representation for molecular systems. The model demonstrates state-of-the-art accuracy in predicting electron density, improving by 30-70% over previous methods when tested on Molecular Dynamics data.
ML for Proteins
Structure-Enhanced Protein Instruction Tuning: Towards General-Purpose Protein Understanding
Proteins are vital biomolecules involved in key biological processes, and accurately predicting their properties is crucial. While protein language models (pLMs) have shown promise with supervised fine-tuning, they are typically tailored to specific tasks, limiting their general-purpose use. To address this, the paper introduces the Structure-Enhanced Protein Instruction Tuning (SEPIT) framework, which integrates structural knowledge into pLMs and connects them to large language models (LLMs) for a more comprehensive understanding of proteins. SEPIT uses a two-stage instruction tuning process: first, it builds a basic understanding of proteins through caption-based instructions, then refines this knowledge with a mixture of experts (MoEs) to learn more complex properties. The framework is trained on the largest protein instruction dataset to date. Experimental results show SEPIT outperforms both open-source and closed-source LLMs in protein-related tasks, demonstrating its effectiveness in general-purpose protein understanding.

Dumpling GNN: Hybrid GNN Enables Better ADC Payload Activity Prediction Based on Chemical Structure
Antibody-drug conjugates (ADCs) are an emerging and highly promising class of targeted cancer therapies, but the design and optimization of their cytotoxic payloads remain a complex challenge. To address this, the study introduces DumplingGNN, a novel hybrid Graph Neural Network (GNN) architecture specifically developed to predict the activity of ADC payloads based on their chemical structure. DumplingGNN integrates Message Passing Neural Networks (MPNNs), Graph Attention Networks (GATs), and GraphSAGE layers, effectively capturing multi-scale molecular features by leveraging both 2D topological and 3D structural information. The model has been evaluated on a comprehensive ADC payload dataset, focusing particularly on DNA Topoisomerase I inhibitors, a key class of cancer drugs. DumplingGNN also demonstrated exceptional performance on various public benchmarks from MoleculeNet, including BBBP (96.4% ROC-AUC), ToxCast (78.2% ROC-AUC), and PCBA (88.87% ROC-AUC). On the specialized ADC dataset, it achieved outstanding accuracy (91.48%), sensitivity (95.08%), and specificity (97.54%). Ablation studies further confirmed that the hybrid GNN architecture, particularly its integration of 3D structural data, was key to enhancing predictive performance.

ML for Omics
De novo design of ATPase based on the blueprint optimized for harboring the P-loop motif
Recent progress in de novo protein design has led to significant insights into protein folding and function. However, the creation of enzymes with activity levels comparable to those found in nature remains a difficult task. In this study, the researchers set out to design an ATPase enzyme from scratch, focusing on identifying an optimal backbone structure that could incorporate a conserved phosphate-binding motif, the P-loop. The designed protein, based on this blueprint, was found to be a monomer with high thermal stability, and it exhibited ATPase activity. Its crystal structure closely aligned with the design model, both in terms of overall structure and the P-loop motif. Interestingly, AlphaFold, a protein structure prediction tool, was unable to accurately predict the folded structure, highlighting the challenges involved in predicting novel protein sequences. Remarkably, the designed protein showed ATPase activity even at temperatures near 100°C, with a notable increase in activity. However, its performance still did not match that of naturally occurring ATPases.

Reviews
Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
LoGG
A long-context RNA foundation model for predicting transcriptome architecture by Ali Saberi
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋