

Portal Weekly #31: mining gene relationships from ChatGPT, identifying perturbation effects, and more.
Portal Weekly #31: mining gene relationships from ChatGPT, identifying perturbation effects, and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬



Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
💻 Latest Blogs
Last week we released a blog post by Austin Tripp arguing that genetic algorithms, a class of methods popular in the 1970s and 80s, are still an important algorithm to keep in mind when doing molecule generation. See his reasons (and especially the takeaways) here!

💬 Upcoming talks
LoGG continues next week with a talk by Mikhail Galkin, who will present ULTRA, an approach for learning universal and transferable graph representations that represents a concrete step towards knowledge graph foundation models.
Join us live on Zoom on Monday, January 22nd at 11 am ET. Find more details here.

We’re still getting things booted up in the new year for the other talk series. Check back on the Events page on Portal for announcements regarding M2D2 and CARE talks.
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
Iterative Prompt Refinement for Mining Gene Relationships from ChatGPT
Large Language Models (LLMs) are sometimes viewed as encoding human knowledge of a domain. One problem is that it can be difficult to tell what outputs from the LLM reflect actual knowledge and what are just hallucinations (not factual). It would be very useful if we could use LLMs to “look up” the relationship between two genes rather than combing through various databases. Toward this goal, this study evaluates GPT-3.5-turbo and GPT-4 abilities to predict gene relationships, benchmarking against the KEGG Pathway Database as “ground truth”. Their evaluations include engaging GPT-4 to suggest improved prompts. “Least-to-most” prompting, deconstructing complex problems into an ordered sequence of manageable subproblems, emerged as a promising technique for their purposes.

ML for Small Molecules
Multi-task ADME/PK Prediction at Industrial Scale: Leveraging Large and Diverse Experimental Datasets
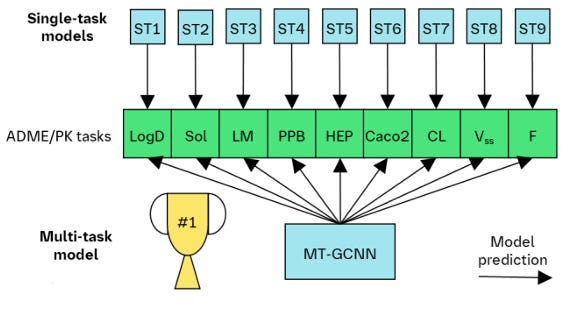
Pharma companies generate a lot of in-house data and don’t have to rely on open-source. This study from Boehringer Ingelheim tested multi-task ML models for predicting ADME and animal PK on in-house endpoints. Their data represent clear lead discovery and optimization trajectories, and they can do realistic time-splits and evaluate their models both at design stage (when they have no experimental data on the test compounds) and at testing stage (when they may have experimental data from earlier assays). They found a clear benefit of using multi-task graph neural networks, and (perhaps unsurprisingly) that endpoints with the largest number of datapoints were especially responsible for increased predictivity.
Graph Learning
Functional MicroRNA-Targeting Drug Discovery by Graph-Based Deep Learning
MicroRNAs are short, single-stranded RNA molecules that modulate different cellular processes and are implicated in several diseases, including cancer. This study applies a graph convolutional neural network strategy and multitask learning to predict molecules that can inhibit specific microRNAs. They used the model to screen nine million molecules for activity against microRNA-21, a driver of breast cancer. They identified a handful of candidate molecules, and experimental follow-up including mouse studies showed that their model is indeed able to successfully suggest compounds that can target specific microRNAs.

ML for Atomistic Simulations
Synthetic Pre-Training for Neural-Network Interatomic Potentials
ML-based models are useful for atomistic materials modeling but depend on the quality and quantity of training data. The idea of “synthetic” (artificial) data is common in other areas of ML research: this study evaluates whether synthetic atomistic data can be used as a pretraining task for neural network interatomic potentials. They show that the approach is feasible and can improve accuracy and computational stability, and do some preliminary tests of the limits of the approach.

ML for Proteins
Pairing Interacting Protein Sequences Using Masked Language Modeling
If we could just look at two amino acid sequences and know whether they interact, we would quickly get a better grasp on many aspects of biology. This study works towards this goal, pairing interacting protein sequences using protein language models like MSA Transformer that are trained on multiple sequence alignments. They suggest that MSA Transformer encodes coevolution patterns between structurally or functionally coupled amino acids, allowing them to uncover structural contacts between proteins. The method apparently works well for small datasets that are otherwise challenging for existing methods.

ML for Omics
A Supervised Contrastive Framework for Learning Disentangled Representations of Cell Perturbation Data
CRISPR technology is allowing us to make targeted gene knockouts in mammalian cells at unprecedented scale, paving the way for a better understanding of gene functions and cell biology. It can be hard to sift through high-dimensional, noisy datasets to determine the sometimes-subtle effects of perturbations, though. This study uses a supervised contrastive variational autoencoder (VAE) that integrates CRISPR guide RNA identity with gene expression data. The authors suggest that the model is sensitive in identifying perturbation effects and also can be used to identify assignment errors and cells escaping the perturbation phenotype.

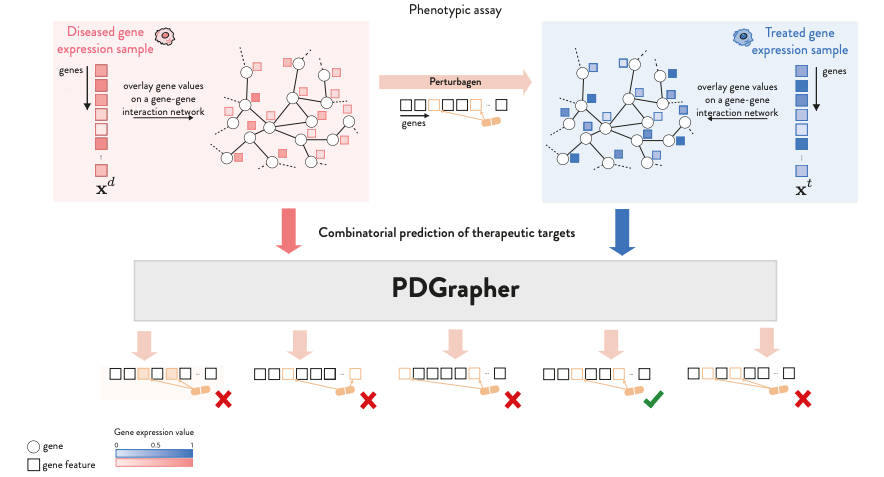
Combinatorial Prediction of Therapeutic Perturbations Using Causally-Inspired Neural Networks
Phenotype-driven approaches identify compounds that counteract the overall disease effects by analyzing phenotypic signatures. This study introduces a graph neural network model, PDGRAPHER, that directly predicts these compounds by learning what kind of perturbations give a specific phenotypic response. Because of the direct approach, PDGRAPHER trains up to 30 times faster. The authors also show on a range of datasets that the model shows improved predictive power and ranking therapeutic targets higher than competing methods.
Open Source
Genomics 2 Proteins Portal: A Resource and Discovery Tool for Linking Genetic Screening Outputs to Protein Sequences and Structures
We can predict many protein structures with high confidence, and we also have loads of high-throughput data that detect protein variants. Mapping the variants onto the structures is useful to understand how variations in protein sequence can cause disease. The G2P portal provides a proteome-wide resource mapping >19 million genetic variants onto ~42,000 protein sequences and ~78,000 structures. It also allows users to interactively upload their own residue-wise annotations and protein structures to establish connections.

Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
LoGG
SUREL+: Moving from Walks to Sets for Scalable Subgraph-based Graph Representation Learning by Haoteng Yin
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋