
Portal Weekly #37: quantum computing for KRAS inhibition, convolutions vs. Transformers, and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬



Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
📅 TechBio Events on the Horizon
Nucleate is launching its Canadian chapter with an AI in BioTech event in Montreal on Wednesday, March 13th. Don’t miss out on an opportunity to hear from various panellists and meet more folks in the TechBio community! Sign up here.

For those of you based in London, Recursion and Valence Labs will be hosting a TechBio mixer in London on Tuesday, March 12th! DM the Valence Labs account to get more details about the event. It seems that there will be an exciting announcement 👀👀👀
💻 Latest Blogs
Last week we had a blog post by Cong Fu and Keqiang Yan giving an overview of LatentDiff, a model combining diffusion models and protein autoencoders to efficiently generate protein structures. Check it out!

💬 Upcoming talks
M2D2 continues next week with a talk by Bowen Jing, presenting a faster way for performing molecular docking by learning a scoring function with a functional form that allows for more rapid optimization.
Join us live on Zoom on Tuesday, March 5th at 11 am ET. Find more details here.

LoGG continues with Ahmed Elhag from Oxford University telling us about Manifold Diffusion Fields (MDF), an approach to learn generative models of continuous functions defined over Riemannian manifolds.
Join us live on Zoom on Monday, March 4th at 11 am ET. Find more details here.

Speakers for CARE talks are usually announced on Fridays after this newsletter goes out. Sign up for Portal and get notifications when a new talk is announced on the CARE events page. You can also follow us on Twitter!
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
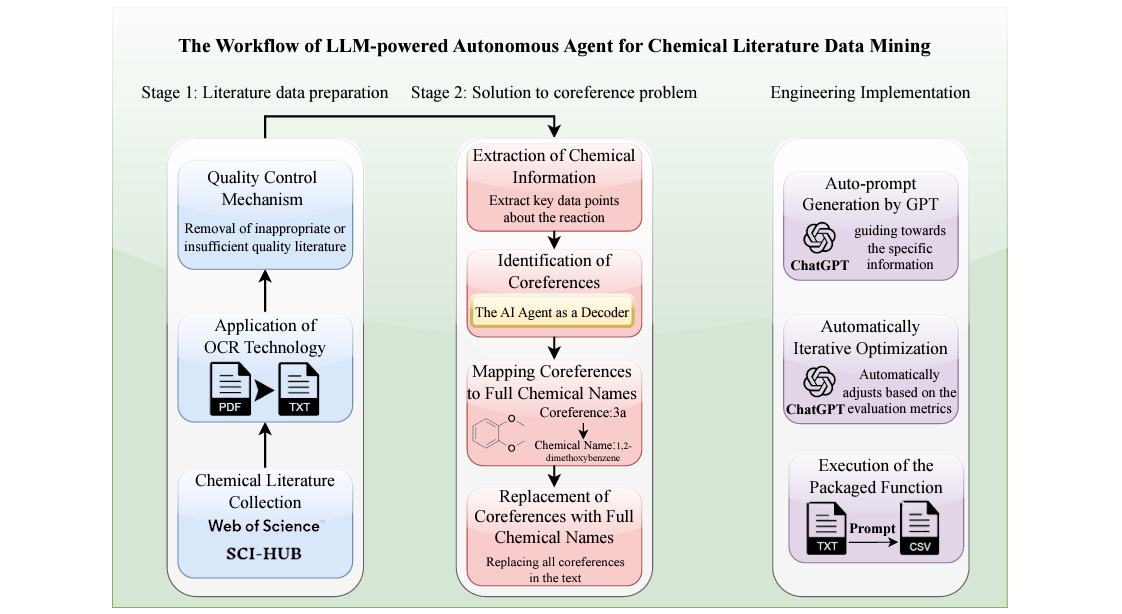
An Autonomous Large Language Model Agent for Chemical Literature Data Mining
Scientific literature is heterogeneous and it’s often hard to find what you’re looking for. This study uses an LLM agent for prompt generation and iterative optimization, mining the literature for information on yield, reactant, catalyst, solvent and product of chemical reactions. They compare their results (evaluated by precision, recall and F1 score) against the performance of ten chemistry graduate students doing manual data collection. On average, the AI agent had competitive precision and much better speed than the humans - it would be interesting to see a follow-up experiment with expert chemists, who are likely faster and more precise but also cost more.
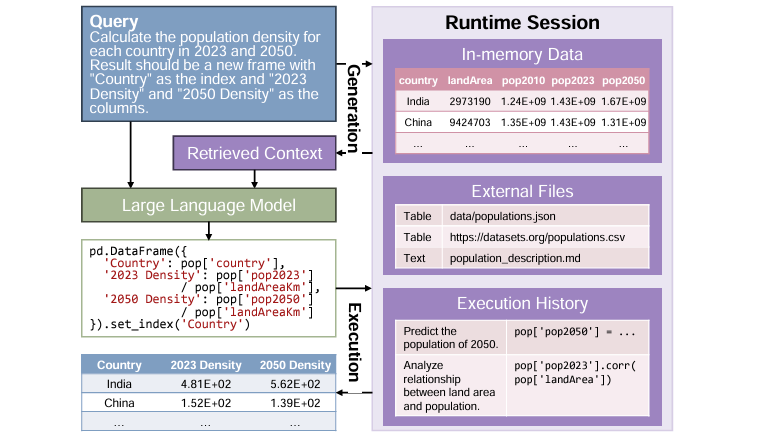
Benchmarking Data Science Agents
LLMs can be useful helpers for data analysis and processing, but real-world applications are challenging and complicated. This study introduces a new evaluation paradigm to assess how well LLM agents perform throughout the entire data science lifecycle. Its findings can help to identify blockers and inform advancements to make the next generation of LLM agents more useful and helpful.
ML for Small Molecules
Quantum Computing-Enhanced Algorithm Unveils Novel Inhibitors for KRAS
It doesn’t get much more cutting-edge than quantum computing. This study uses quantum algorithms trained on a 16-qubit IBM quantum computer, combined with established models for designing small molecules. They apply their model to design new inhibitors of KRAS (an oncogene that drives cancer progression). Of the 15 molecules they synthesized during their study, two were experimentally confirmed as binding to KRAS at low micromolar concentrations, providing an applied example of quantum computing in drug discovery.

Adapting Deep Learning QSPR Models to Specific Drug Discovery Projects
Global ML models in chemistry are usually trained on large datasets with diverse chemical matter, and are used as a general solution in drug discovery projects. Local models can be used during specific projects to focus on more targeted parts of chemical space. This study benchmarks global, local, and hybrid models on more than 300 drug discovery projects and 10 ADMET assays at Novartis. Overall, fine-tuning a pre-trained global ML model performed best, providing support for the idea that transfer learning (domain adaptation) is a promising strategy in molecular property prediction.

ML for Atomistic Simulations
MFBind: a Multi-Fidelity Approach for Evaluating Drug Compounds in Practical Generative Modeling
Generative models for drug discovery use molecular docking to evaluate the quality of generated compounds, but high docking scores do not necessarily translate to experimental activity (as the recent CACHE hit-finding competition shows!). There is often a tradeoff between accuracy and computational cost in modeling. This study proposes a multi-fidelity approach, integrating docking and binding free energy simulations to train a deep surrogate model with active learning. They use an environment with a set of simulators that output an increasingly accurate estimate of the value of interest and try to balance tradeoffs. This approach seems to be able to do better than single-fidelity methods and effectively use a limited computational budget, which is useful if you’re more resource-constrained.

ML for Proteins
Convolutions are Competitive With Transformers for Protein Sequence Pretraining
Protein language models (PLMs) can be powerful, but since they rely on Transformer architecture they may have trouble modeling longer-range interactions and scale quadratically because of the attention mechanism. Convolutions can be powerful because they scale linearly with sequence length. This study compares the two approaches in PLMs, and finds that with masked language model pretraining, convolutional neural network (CNN) architectures can be competitive to (and sometimes better than) Transformers across a range of downstream applications. This suggests that sometimes you can swap out a Transformer for a CNN and get better computational efficiency. It also means we might need to disentangle pretraining task and model architecture so we know where performance gains are coming from.

Machine Learning Approaches in Predicting Allosteric Sites
Allostery is when binding at one side of a protein affects the binding activity at another functional site. This can be useful if you want a noncompetitive inhibition, for example; your drug doesn’t need to fight the native ligand to get into the active site. This review gives an overview of ML models that have been developed to predict allosteric sites, and gives a perspective on the field.

ML for Omics
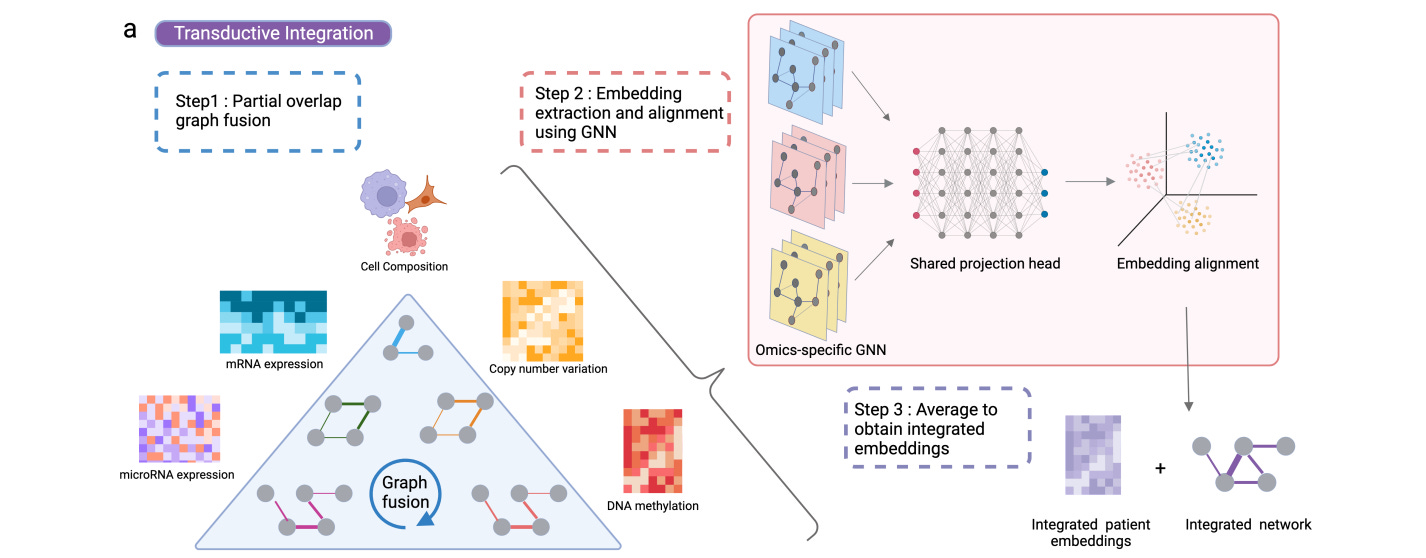
Integrate Any Omics: Towards Genome-Wide Data Integration for Patient Stratification
Although multi-omics is likely to be more powerful for models than any individual modality, sometimes the data are patchy: we may only have partial omics data for some samples. Sample exclusion risks throwing away information, and imputation can potentially lead the model astray or fail to reflect biological diversity. This study attempts to combine incomplete multi-omics data using partially overlapping patient (sample) graphs and graph neural networks. Their results, using six omics modalities across five cancer cohorts, suggest that their approach gives robustness to missing data and increases accuracy in classifying new samples with partial profiles.
Open Source
CMD + V for chemistry: Image to Chemical Structure Conversion Directly Done in the Clipboard
Experts can look at a molecule and know exactly what it’s called, but for the rest of us Clipboard-to-SMILES Converter tries to provide an easy way to convert from images to common molecular representations. It also incorporates SMILES operations including canonicalization and price-searching of molecules, and can continuously monitor the clipboard and automatically convert supported representations or images into SMILES.

Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
M2D2
Multiflow: protein structure and sequence co-generation through generative flows on discrete state-spaces by Jason Yim and Andrew Campbell
LoGG
AlphaFold Meets Flow Matching for Generating Protein Ensembles by Bowen Jing
CARE
BISCUIT: Causal Representation Learning from Binary Interactions by Phillip Lippe
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋