


Portal Weekly #41: guided diffusion for molecule generation, large antibody language models, and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬



Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
We’re going to do something different next week and feature a member of the Portal TechBio community! Chaitanya K. Joshi is a PhD student in the Department of Computer Science at the University of Cambridge, supervised by Prof. Pietro Liò.

We’re planning to ask him about his journey into ML for molecular systems for protein and RNA design, what it’s like doing research at Cambridge, his main takeaways from his recent Hitchhiker's Guide to Geometric GNNs for 3D Atomic Systems (see below!) and gRNAde: Geometric Deep Learning for 3D RNA inverse design. If you have questions you’d like us to ask, please fill out this form!
💻 Latest Blogs
This week we had a blog post by Kyle Swanson introducing ADMET-AI, a fast and accessible platform for predicting absorption, distribution, metabolism, excretion, and toxicity properties. The model has the best average performance on TDC and boasts a web interface as well as a Python package. Kyle walks you through the interface and model here - check it out!

💬 Upcoming talks
M2D2 continues next week with a talk by Emma Svensson, who will tell us about HyperPCM a method that uses HyperNetworks to efficiently transfer information between tasks during inference for better generalization in predicting drug-target interactions.
Join us live on Zoom on Tuesday, April 2nd at 11 am ET. Find more details here.

LoGG continues with a presentation by Simon Mathis, Chaitanya Joshi and Alexandre Duval, who will be talking about their recent paper (self-described as “opinionated”!), a survey on Geometric GNNs. This talk is likely to have something for newcomers and more experienced folks, so tune in!
Join us live on Zoom on Monday, April 1st at 11 am ET. Find more details here.

Speakers for CARE talks are usually announced on Fridays after this newsletter goes out. Sign up for Portal and get notifications when a new talk is announced on the CARE events page. You can also follow us on Twitter!
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
Large Scale Paired Antibody Language Models
Antibodies are incredibly diverse: they need to be, to be able to bind with high specificity to foreign substances like bacteria, viruses or toxins and alert the immune system. With modern technologies, we can amass huge amounts of antibody sequences, but using these data to design better therapeutics is hampered by the size and complexity of the data. This study presents two antibody-specific language models, trained on more than two billion sequences from the Observed Antibody Space dataset. These models seem to do better than existing antibody and protein language models on a range of antibody-engineering tasks but may do worse than general protein models at predicting protein expression, demonstrating tradeoffs in LLM specialization.

Bioinformatics and Biomedical Informatics with ChatGPT: Year One Review
ChatGPT has been around for about a year, and it’s had different effects on different industries and domains. This review looks at how it has been used in omics, genetics, biomedical text mining, drug discovery and more. They try to identify the current strengths and limitations of ChatGPT and suggest areas for future improvement.

ML for Small Molecules
SILVR: Guided Diffusion for Molecule Generation
A Journal of Chemical Information and Modeling Editor’s Choice article, this study proposes a method to condition a diffusion-based equivariant generative model without retraining it. Using fragment hits (a molecule of low molecular weight that has been validated to bind to a target protein), SILVR can generate new molecules with a similar shape to the originals, meaning that they will likely fit the binding site without having to have direct knowledge of the protein, and can also merge up to 3 fragments into a new molecule. As long as you have fragment data and a diffusion-based generative model, this method can be useful for designing new potential drug-like molecules.

Leap: Molecular Synthesisability Scoring with Intermediates
For a drug to be effective (or even tested), it first has to be made. Synthesis-planning algorithms can be slow and hard to combine with generative methods that try to assess large numbers of molecules. This study suggests a synthesizability scoring method, Leap, that pre-trains a GPT-2 model to predict the entire multi-step retrosynthesis route for a target compound and then fine-tunes using a retrosynthetic planning tool to predict a model’s synthetic complexity. They argue that this approach allows for a more nimble model that can adapt to the presence of useful intermediates for a target molecule.
Interested in learning more about synthesis planning and its pitfalls? Austin Tripp gave an excellent talk on the subject at M2D2 recently - check it out!

ML for Atomistic Simulations
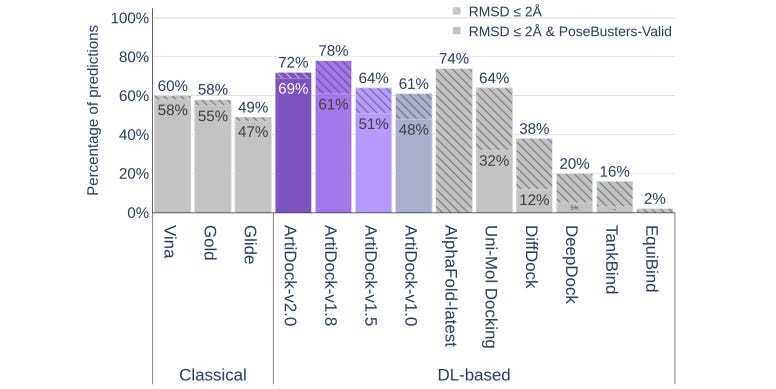
ArtiDock: Fast and Accurate Machine Learning Approach to Protein-Ligand Docking Based on Multimodal Data Augmentation
Data augmentation has been useful in computer vision - adding rotated, flipped or polarized images can help a model learn more robust representations. It’s particularly useful when you’re limited in training data. This study augments ML docking with artificial binding pockets and ensembles of representative conformations of ligand-protein complexes from MD simulations. They train a fairly simple model that seems to do well on the PoseBusters benchmark task and is significantly faster than its closest competitors, suggesting that some augmentation may be useful in this domain.
What is the PoseBusters benchmark, and why should we use it? Martin Buttenschoen gave an M2D2 talk introducing it! Check out the talk and the paper here!
ML for Proteins
Engineering of Highly Active and Diverse Nuclease Enzymes by Combining Machine Learning and Ultra-High-Throughput Screening
Highly autonomous agents will one day be able to perform science and extract nobel-worthy insights, but in the meantime, ML can be effectively used to guide multi-round discovery campaigns. This study aims to engineer a nuclease with applications in the treatment of chronic wounds. Models trained on evolutionary data alone, without access to any experimental data, designed functional variants at a significantly higher rate than a traditional approach to initial library generation. ML-designed hits were more diverse and active than those from traditional directed evolution campaigns that were run in parallel. The authors also provide a dataset of 55K diverse variants to help in future studies of enzyme activity landscapes.

ML for Omics
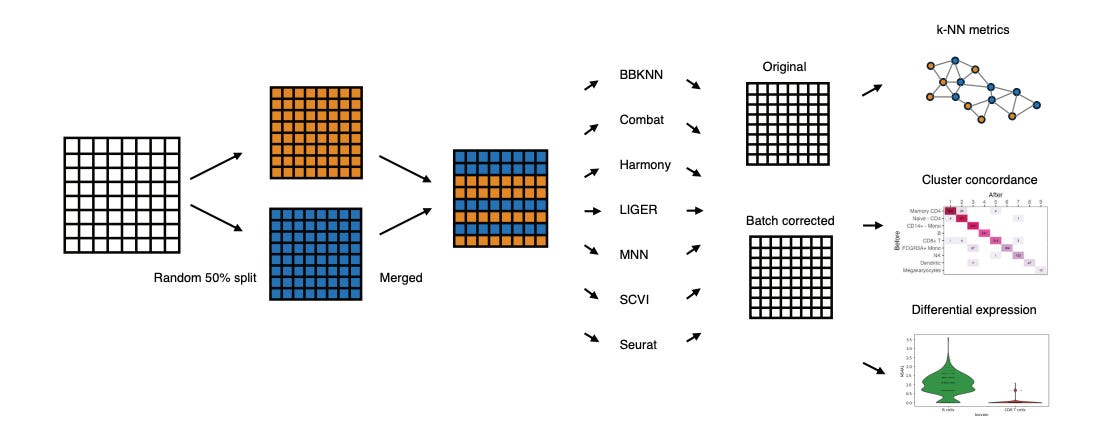
Batch Correction Methods Used in Single Cell RNA-Sequencing Analyses are Often Poorly Calibrated
We always have the desire to combine data from different experiments to bump up our dataset size, but we have to worry about batch effects when we do that. This study compares seven widely used methods for batch correction of scRNA-seq datasets. Out of MNN, SCVI, LIGER, Combat, BBKNN, Seurat and Harmony, the authors found that Harmony was the only one that consistently performed well in their tests (kNN metrics, cluster concordance and differential expression looking at two batches randomly split from the same dataset). This is useful information since pre-processing and standardization are major components of good omics analyses.
Batch effects are troublesome, and it’s important to compare different approaches for combating them. Karin Hrovatin discusses some batch correction strategies: check out her takeaways here! You might also be interested in Chenyu Wang’s talk on addressing batch effects with Information Maximization.
Open Source
Lit-OTAR Framework for Extracting Biological Evidences from Literature
The biomedical literature is massive and it can be hard to find the interactions that you care about. This effort uses Named Entity Recognition (NER) for identifying genes/proteins, diseases, organisms, and chemicals/drugs within scientific texts, and entity normalization to map these entities to databases like Ensembl, Experimental Factor Ontology (EFO), and ChEMBL. They say that they’ve processed 39 million abstracts and 4.5 million full-text articles so far. They make the results accessible through the Open Targets Platform and the Europe PMC website.

Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
M2D2
Diffusion Models on Sampling Rare Events by Chenru Duan
LoGG
MFBind: a Multi-Fidelity Approach for Evaluating Drug Compounds in Practical Generative Modeling by Peter Eckmann
CARE
Meaningful Causal Aggregation and Paradoxical Confounding by Yuchen Zhu
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋