


Portal Weekly #50: a GUI for feedback from chemists, first principles accuracy in alchemical ML, a review on protein structure and docking diffusion models and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬



Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
💻 Latest Blogs
What’s better than diffusion for ligand design? Diffusion with expert knowledge. In the latest blog, Yael Ziv introduces MolSnapper, a tool designed to condition diffusion models for structure-based drug design. Check it out here.

Simon Geisler introduces S²GNNs, which generalize the spatial + FFT convolution/global operation of sequence models like H3/Hyena/Mamba to graphs, giving an efficient model that can compete with graph transformers and MPGNNs. Check it out here. (Simon gave a LoGG talk last year on transformers in directed graphs - check it out)

Single-cell biology is extremely complex, and large foundation models may contribute to a solution. Abhishaike Mahajan gives an intro to scRNA-seq, cell atlases, and foundation models in the latest blog. Check it out here.

💬 Upcoming talks
M2D2
Next week, Chaitanya Joshi will introduce gRNAde, a geometric RNA design pipeline for the 3D inverse folding problem (given a structure, predict what sequence could fold into that structure). The model recovers native sequences at a higher rate than Rosetta, while making it predictions in seconds rather than hours.
Join us live on Zoom on Tuesday, June 11th at 11 am ET. Find more details here.

Speakers for CARE talks are usually announced on Fridays after this newsletter goes out. Sign up for Portal and get notifications when a new talk is announced on the CARE events page. You can also follow us on Twitter and LinkedIn!
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
Large Property Models: A New Generative Paradigm for Molecules
Generative models for molecule design may suffer when data are scarce. This paper suggests that a generative model trained on multiple ‘accessible molecular properties’ like compound complexity and dipole moment would show an accuracy phase transition after achieving a sufficient size, similar to what has been seen with large language models. They train transformers to take molecular properties and predict a molecular graph. This is an early stage of the study, but represents a different paradigm for molecular language models.

ML for Small Molecules
Metis - A Python-Based User Interface to Collect Expert Feedback for Generative Chemistry Models
You can have a great ML model, but without adoption it’s useless. With the goal of aligning models’ outputs to align with chemists’ preferences, this preprint presents an open-source graphical user interface to collect detailed feedback on molecular structures. This tool should allow ML researchers to more effectively capture chemists’ implicit knowledge and preferences, and to help ML models make a bigger impact in real drug discovery campaigns.

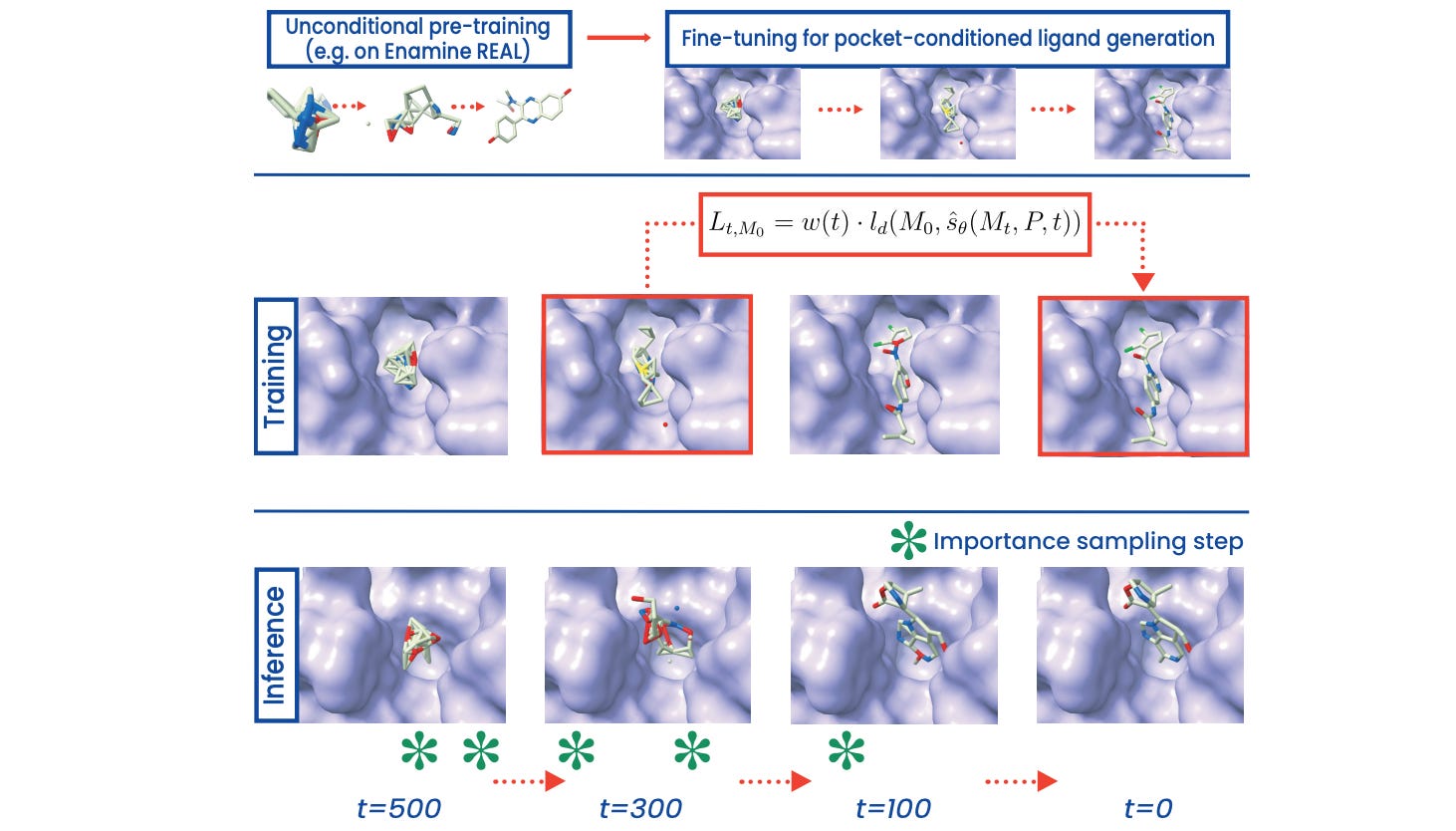
PILOT: Equivariant diffusion for pocket conditioned de novo ligand generation with multi-objective guidance via importance sampling
It’s difficult to design molecules with desired properties that fit a given protein pocket. This study from Pfizer introduces an equivariant diffusion model with a sampling strategy designed to direct the molecule towards increased binding affinity and synthetic accessibility. The model performs well on CrossDocked 2020, and over 98% of molecules were PoseBusters-valid.
ML for Atomistic Simulations
Computing hydration free energies of small molecules with first principles accuracy
A large fraction of the computation performed in pharmaceutical R&D is free energy calculations, and alchemical calculations can help simplify the problem. This study introduces an efficient alchemical free energy method compatible with machine learned forcefields, enabling calculation of hydration free energies with first principles accuracy. They use a soft-core MACE architecture and provide an implementation of the method through OpenMM.

ML for Proteins
Cramming Protein Language Model Training in 24 GPU Hours
State-of-the-art protein language models like ESM2 take hundreds of thousands of GPU hours to pre-train on the vast protein universe. This study describes ways of training a 67 million parameter model in a single day - the model gets similar performance on downstream protein fitness landscape inference tasks as ESM-3B, despite being trained for > 15000 more GPU hours. The authors provide an open-source model training and inference library, with the catchy acronym of LBSTER.

AlphaFold3, a secret sauce for predicting mutational effects on protein-protein interactions
AlphaFold3 is the new state-of-the-art, and everyone wants to see how good it is. This work looks at how AF3 predictions compare or synergize with other deep learning (DL) methods in binding energy prediction. They found that AF3 was fairly good (better than other DL methods) but still worse than classic force fields. Ensembling AF3 and force field predictions could yield something better than either, since they are orthogonal techniques.

ML for Omics
CELLama: Foundation Model for Single Cell and Spatial Transcriptomics by Cell Embedding Leveraging Language Model Abilities
This study proposes a framework that uses techniques from language modeling to transform cell data include gene expression and metadata into ‘sentences’ using a sentence transformer. Embeddings derived from this technique were used in a range of downstream tasks, circumventing dataset-specific analytical workflows.

Open Source
JANE - Journal/Author Name Estimator
“Have you recently written a paper, but you're not sure to which journal you should submit it? Or maybe you want to find relevant articles to cite in your paper? Or are you an editor, and do you need to find reviewers for a particular paper? Jane can help! JANE relies on the data in PubMed, which can contain papers from predatory journals, and therefore these journals can appear in JANE's results. To help identify high-quality journals, JANE now tags journals that are currently indexed in MEDLINE, and open access journals approved by the Directory of Open Access Journals (DOAJ).”

Reviews
Diffusion models in protein structure and docking
Diffusion models (DM) are well equipped to model high dimensional, geometric data while exploiting key strengths of deep learning. This review covers the basics of diffusion models, modeling choices regarding molecular representations, generation capabilities, prevailing heuristics, and key limitations and forthcoming refinements. They provide best practices around evaluation procedures to help establish rigorous benchmarking and evaluation.

Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
LoGG
The Continuous Language of Protein Structure by Ben Murrell
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋