


Portal Weekly #59: LipidBERT, SMILES-Mamba, DNA sequencing data, GPUs, and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬
Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
💬 Upcoming talks
LoGG continues next week with a talk by Andre Cornman, co-founder of Tatta Bio, a non-profit startup building genomic intelligence. He’ll be able to answer all your questions about the OMG dataset.
Join us live on Zoom on Monday, August 26th at 11 am ET. Find more details here.

Speakers for CARE talks are usually announced on Fridays after this newsletter goes out. Sign up for Portal and get notifications when a new talk is announced on the CARE events page. You can also follow us on Twitter and LinkedIn!
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
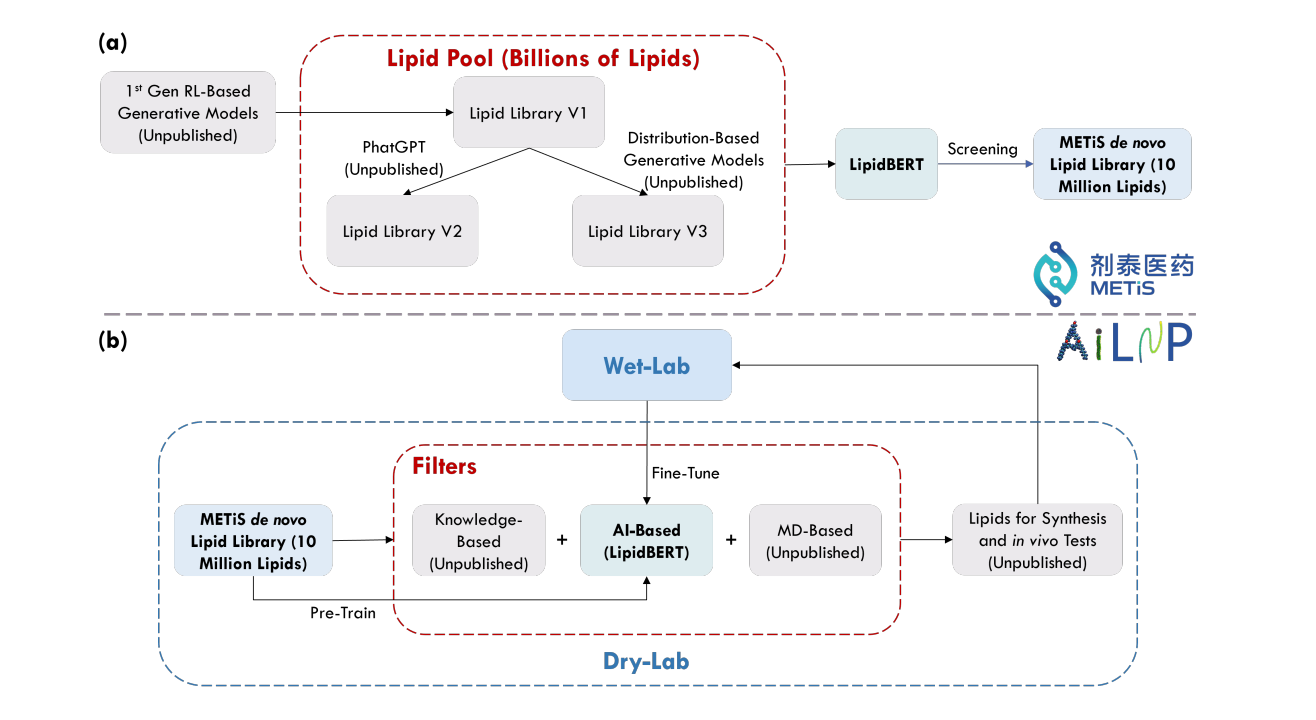
LipidBERT: A Lipid Language Model Pre-trained on METiS de novo Lipid Library
This paper looks at generating and maintaining a 10 million virtual lipids database through METiS's in-house de novo lipid generation algorithms and lipid virtual screening techniques. They also compare the performance of embeddings generated by LipidBERT and PhatGPT (GPT-like lipid generation model). It’s the first successful demonstration of the capability of a pre-trained language model on virtual lipids and its effectiveness in downstream tasks using web-lab data!
ML for Small Molecules
SMILES-Mamba: Chemical Mamba Foundation Models for Drug ADMET Prediction
Predicting absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of small-molecule drugs is often resource-intensive and requires extensive experimental data. Enter SMILES-Mamba, a two-stage model that leverages unlabeled and labeled data through self-supervised pretraining and fine-tuning strategies. SMILES-Mamba achieves the highest score in 14 tasks across 22 ADMET datasets, highlighting the potential of self-supervised learning in improving molecular property prediction.
ML for Atomistic Simulations
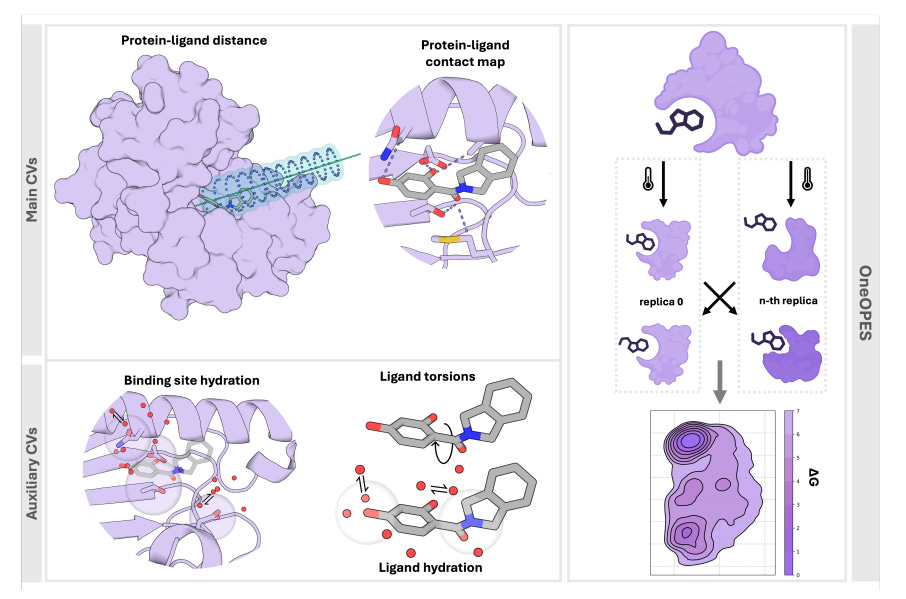
Absolute Binding Free Energies with OneOPES
Absolute binding free energy (ABFEs) calculations have been improving but achieving the level of accuracy required to inform drug discovery efforts remains difficult. This paper presents an enhanced sampling strategy to accurately calculate absolute binding free energies using OneOPES with simple geometric collective variables. It effectively samples 15 different ligand binding modes and consistently matches the experimentally determined structures regardless of the initial protein-ligand configuration.
ML for Proteins
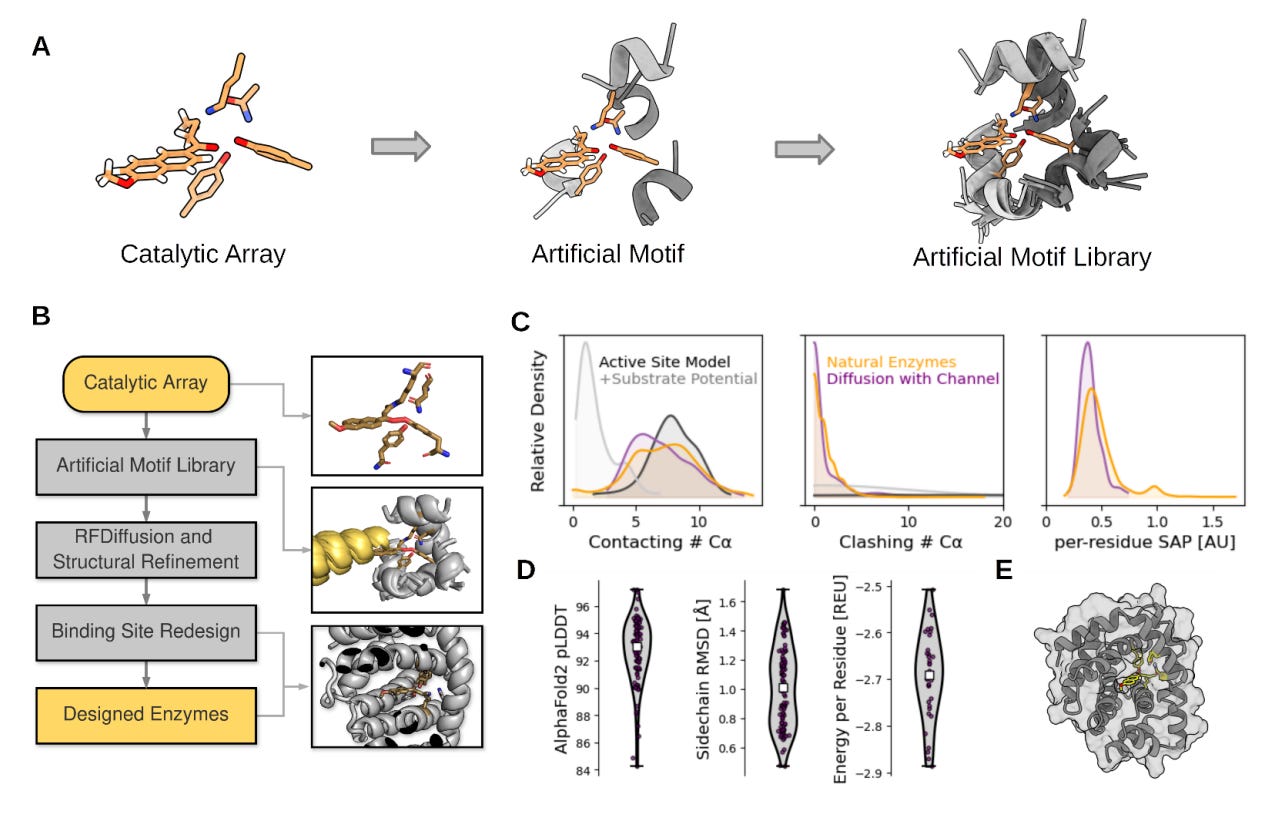
Computational design of highly active de novo enzymes
In the world of enzymes, custom-designing enzymes can help boost the use of biocatalysts in industrial biotransformations. This paper reviews rotamer inverted fragment finder – -diffusion (Riff-Diff), a hybrid machine learning and atomistic modeling strategy for scaffolding catalytic arrays in de novo protein backbones with custom substrate pockets. It claims that in principle, their design can be applied to any catalytically competent amino acid constellation.
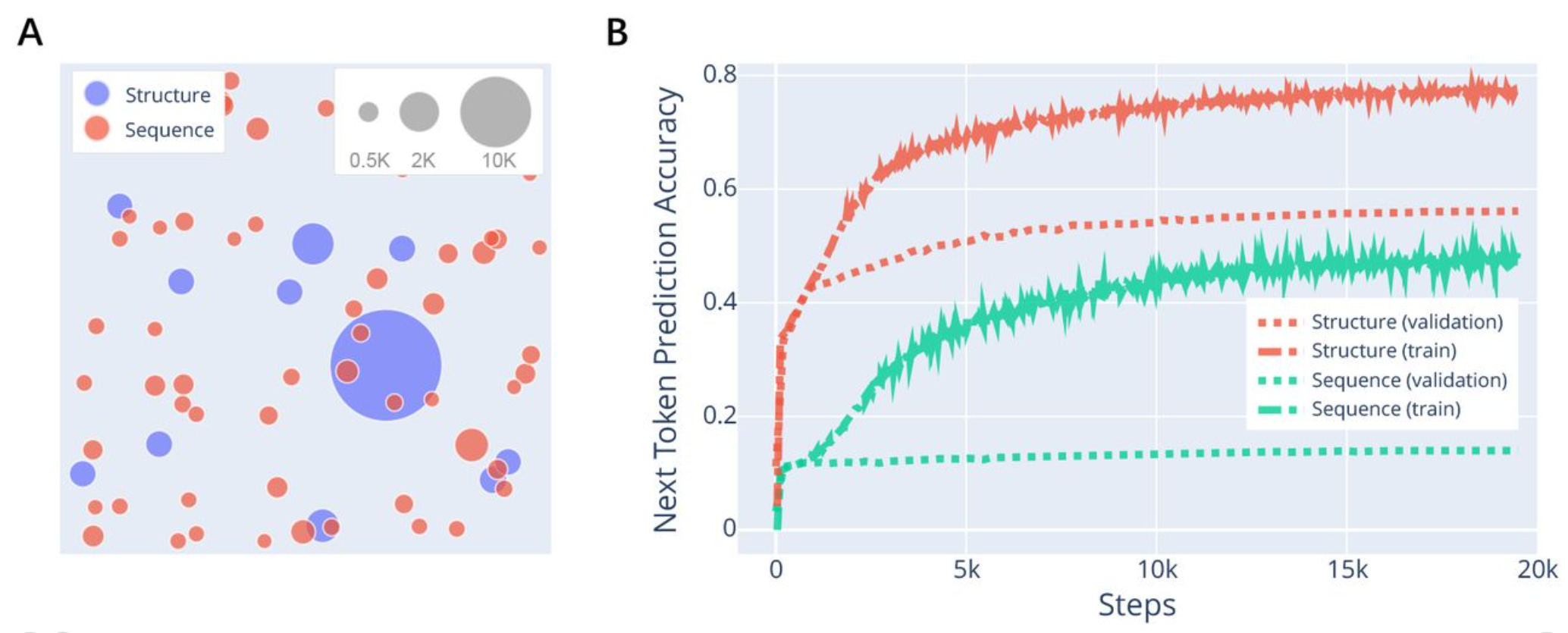
Toward De Novo Protein Design from Natural Language
De novo protein design (DNPD) aims to create new protein sequences from scratch, without relying on existing protein templates. Current methods are often too specific or too narrow on defined protein designs. Pinal, a probabilistic sampling method that generates protein sequences using rich natural language as guidance employs a two-stage generative process. Results show that Pinal outperforms existing models, including the concurrent work ESM3.
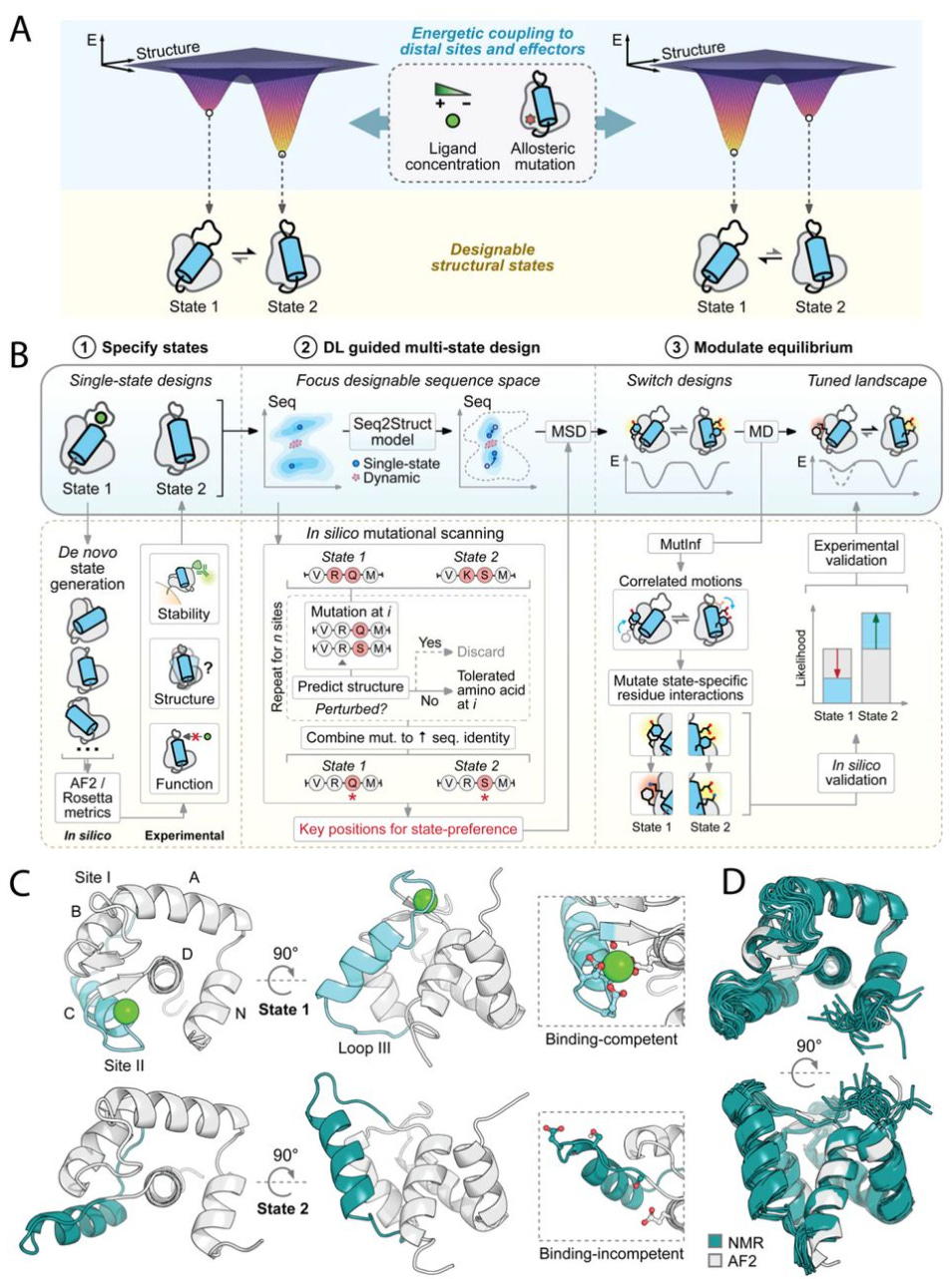
Deep learning guided design of dynamic proteins
Controlled conformational dynamics are hallmarks of natural switch-like signaling proteins and have remained mostly inaccessible to de novo design. This paper presents a general deep-learning-guided approach for de novo design of dynamic changes between intra-domain geometries of proteins, similar to switch mechanisms prevalent in nature, with atom-level precision. They show that new modes of motion can now be realized through de novo design, and provide a framework for constructing biology-inspired, tunable and controllable protein signaling behavior de novo.
ML for Omics
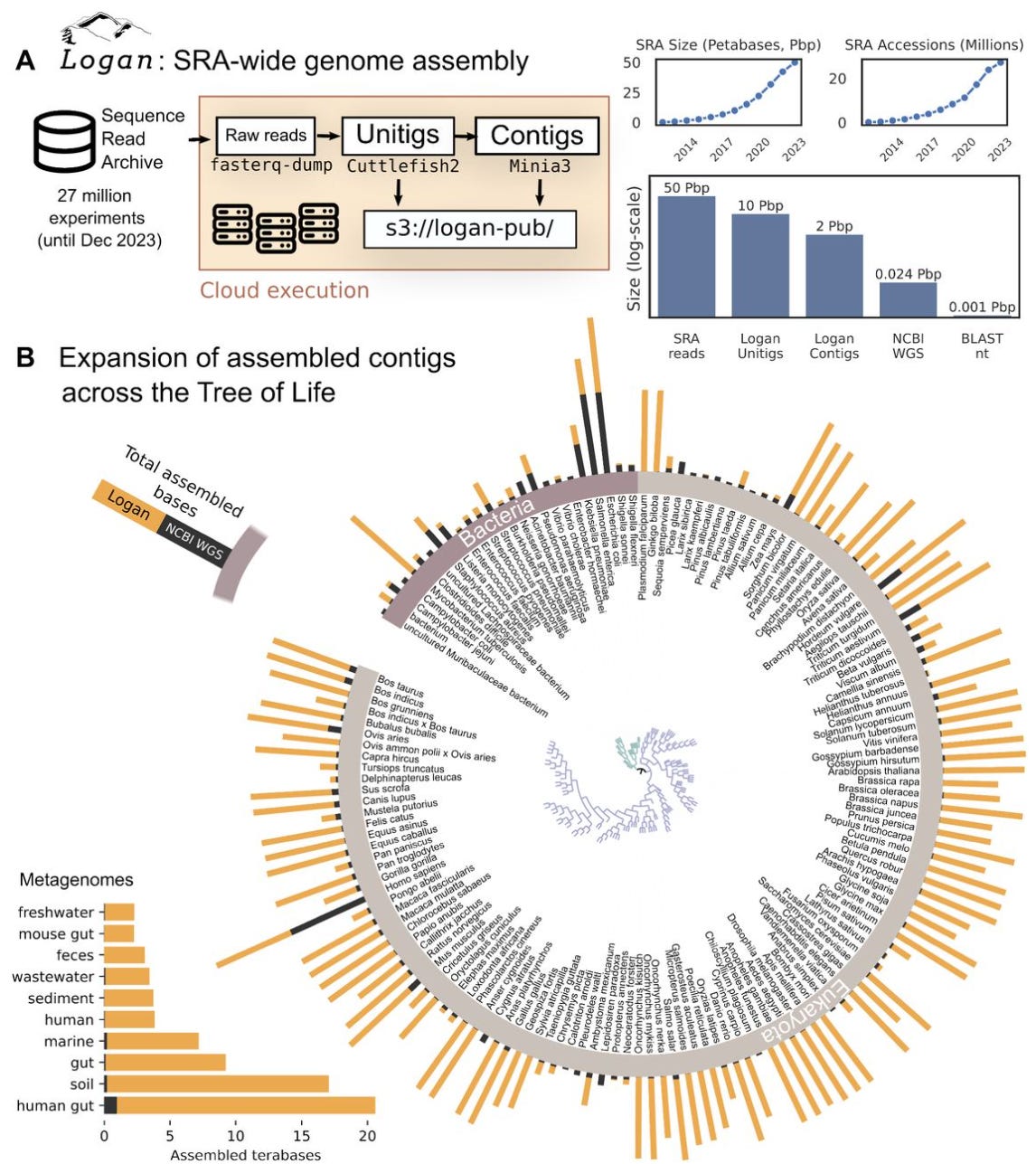
Logan: Planetary-Scale Genome Assembly Surveys Life’s Diversity
Did you know that the NCBI Sequence Read Archive (SRA) is the largest public repository of DNA sequencing data? It contains the most comprehensive snapshot of Earth’s genetic diversity to date. Now imagine searching through the entire database of almost everything on Earth for genetic sequences of interest in a reasonable time. The logan assemblage is the largest dataset of assembled sequencing data to date and aims to enable a new era of accessible petabase-scale computational biology inquiry. Results claim logan can complete queries in as little as 11 hours. Happy searching!
Open Source
Clustering one million molecular structures on GPU within seconds
If you thought searching through DNA sequencing data was large, structure clustering might come close. Most tools cannot support the clustering analysis on GPUs focusing on the root mean square deviation metric. This paper introduces a highly efficient GPU-accelerated tool for molecular structure clustering. On two NVIDIA RTX4090 GPUs and single precision data type, the clustering calculation on 1 million snapshots is completed in a few seconds, which is hundreds of times faster than on CPUs.
Reviews
Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
LoGG
Tokenized and Continuous Embedding Compressions of Protein Sequence and Structure by Amy Lu
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋