
Portal Weekly #67: MoML 2024, data-driven discovery, boltzmann-aligned inverse folding models, geometric contexts, and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬



Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
⭐ Polaris Launches New Steering Committee
Polaris is excited to share the launch of its Steering Committee, bringing together a group of industry experts dedicated to developing guidelines for benchmarking best practices - starting with small molecules.
Their first publication in Nature Machine Intelligence dives into common challenges in benchmarking and emphasizes the need for collaboration across industries, helping to connect innovators and practitioners in meaningful ways. Plus, they’ll soon be releasing a pre-print on method comparison to improve reproducibility in drug discovery.

For more details, check out their announcement.
🗓️ MoML @ MIT 2024
The Molecular Machine Learning (MoML) Conference 2024 is back and will be held on November 5 at the Koch Institute for Integrative Cancer Research at MIT in Cambridge, MA. This event gathers researchers from academia and industry to explore advancements in machine learning for molecular modeling and therapeutic design. Attendees can expect engaging talks, poster presentations, and best paper prizes. Limited free admission is available for non-MIT students with accepted papers.
For more details, visit MoML 2024.

💬 Upcoming talks
LoGG continues next week with a talk by Lazar Atanackovic to discuss modeling the continuous evolution of interacting biological systems using Meta Flow Matching (MFM). He’ll be able to answer all your questions. Please note that LoGG has changed from 11 am ET to 12 pm ET.

Join us live on Zoom on Monday at 12 pm ET. Find more details here.
Speakers for CARE talks are usually announced on Fridays after this newsletter goes out. Sign up for Portal and get notifications when a new talk is announced on the CARE events page. You can also follow us on Twitter and LinkedIn!
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
InstructBioMol: Advancing Biomolecule Understanding and Design Following Human Instructions
Understanding and designing biomolecules like proteins and small molecules is crucial for advancing fields such as drug discovery, synthetic biology, and enzyme engineering. While large language models (LLMs) have shown promise in interpreting human intentions, their use in research still faces challenges like specialized knowledge requirements and the need to integrate various types of data. This study introduced InstructBioMol, a novel LLM that bridges the gap between natural language and biomolecules by allowing for a comprehensive alignment of language with molecular structures. In experiments, InstructBioMol demonstrated its ability to understand and design biomolecules based on human instructions, generating drug molecules with a 10% improvement in binding affinity and creating enzymes that achieved an impressive ESP Score of 70.4.
Large language models and their applications in bioinformatics
Recent advancements in Natural Language Processing (NLP) have been largely fueled by the rise of Large Language Models (LLMs), which represent a major leap in language technology. Beyond traditional NLP uses, LLMs are making significant strides in bioinformatics, helping to tackle challenges related to large and complex biological datasets. In areas like genomics, proteomics, and personalized medicine, LLMs aid in identifying patterns, predicting protein structures, and understanding genetic variations—capabilities that are vital for advancing drug discovery and accurately predicting molecular interactions.
ML for Small Molecules
The future of machine learning for small-molecule drug discovery will be driven by data
Small-molecule therapeutics make up about 90% of all approved medicines and are widely used in the pharmaceutical industry because they can easily penetrate cells and target specific proteins or RNA. But developing these drugs is really tough and expensive, with long timelines and high failure rates. There’s hope that machine learning (ML) could speed up the process and lower costs, just like it’s revolutionized other fields like computer vision and protein structure prediction. However, ML hasn’t had the same breakthrough in small-molecule drug discovery yet because the models often don’t perform well when applied to new data. While there’s been progress, the field is still waiting for its “AlphaFold moment” – a big leap forward like we’ve seen with protein structure prediction.

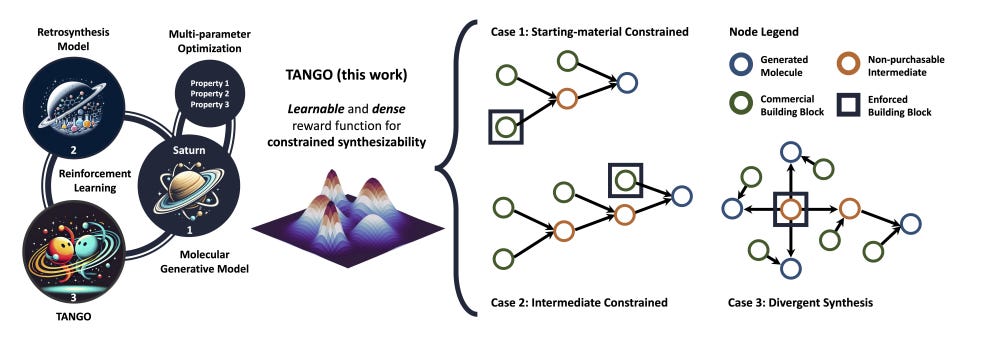
It Takes Two to Tango: Directly Optimizing for Constrained Synthesizability in Generative Molecular Design
Constrained synthesizability is a significant but often overlooked challenge in generative molecular design, particularly when it comes to creating molecules that not only meet multiple optimization objectives but are also synthesizable using specific commercial building blocks. In this study, researchers introduced a new reward function called Tanimoto Group Overlap (TANGO), which leverages principles of chemistry to turn a sparse reward function into a dense, learnable one—essential for reinforcement learning. TANGO enhances general-purpose molecular generative models, allowing them to directly optimize for synthesizability while also considering other important properties in drug discovery.
ML for Atomistic Simulations
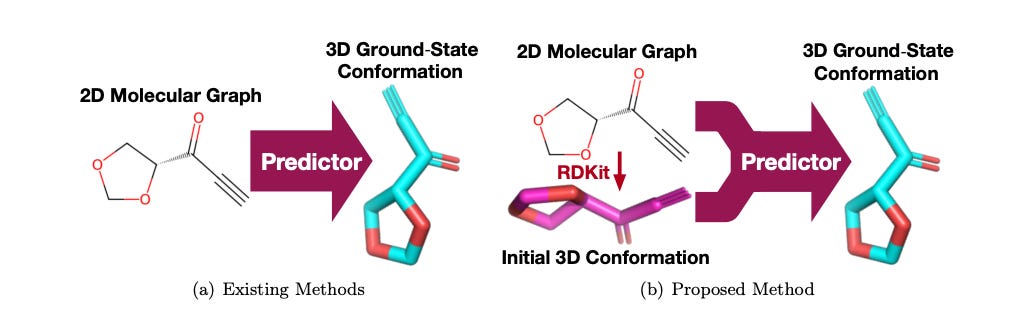
Predicting Molecular Ground-State Conformation via Conformation Optimization
Predicting the ground-state conformation of molecules from their molecular graphs is important for various chemical applications like molecular modeling and docking. Recently, many learning-based methods have emerged to replace the slow simulations typically used for this task, but they often fall short because they only use the molecular graph data to make predictions from scratch. ConfOpt works by iteratively refining the low-quality conformation with guidance from the molecular graph, optimizing the atomic 3D coordinates and the distances between atoms simultaneously during training. Extensive tests show that ConfOpt performs significantly better than existing methods, offering a more efficient and accurate way to predict molecular ground-state conformations.
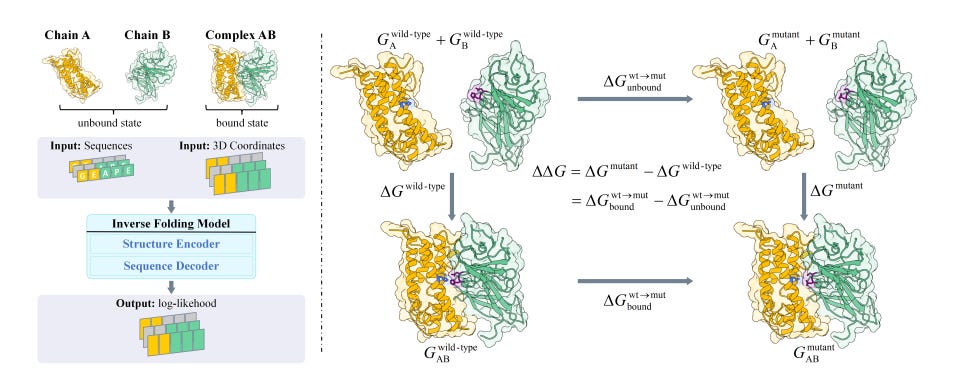
Boltzmann-Aligned Inverse Folding Model as a Predictor of Mutational Effects on Protein-Protein Interactions
Predicting changes in binding free energy (∆∆G) is essential for understanding and modifying protein-protein interactions, which are important in drug design. Since experimental ∆∆G data is limited, most existing methods focus on pretraining but often overlook the significance of aligning data. To address this, researchers have introduced a new technique called Boltzmann Alignment, which transfers knowledge from pre-trained inverse folding models to improve ∆∆G predictions. In experiments with the SKEMPI v2 dataset, they achieved impressive Spearman coefficients of 0.3201 for unsupervised and 0.5134 for supervised predictions, far surpassing previous records.
ML for Proteins
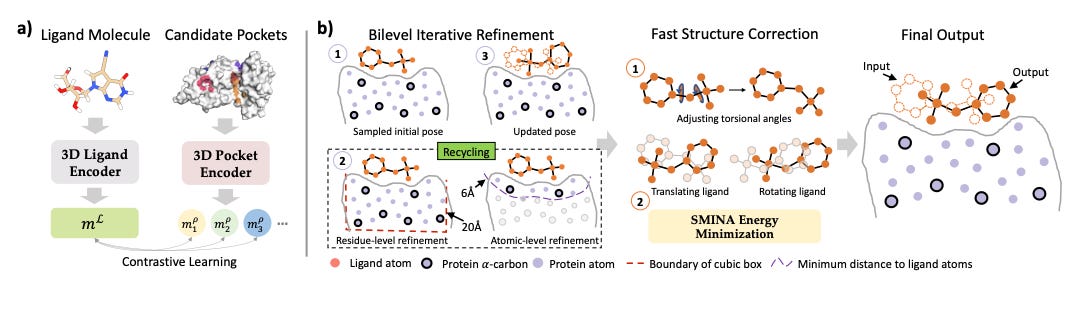
DeltaDock: A Unified Framework for Accurate, Efficient, and Physically Reliable Molecular Docking
Molecular docking is a key technique in drug design that helps predict how ligands bind to proteins, which is essential for understanding protein-ligand interactions. However, many current methods are designed for specific scenarios and struggle with issues like ignoring protein side chains or dealing with large binding pockets. To address these challenges, researchers developed a new two-stage docking framework called DeltaDock, which includes pocket prediction and site-specific docking. DeltaDock uses a detailed iterative refinement process for more accurate docking, and experiments show that it significantly outperforms previous models, achieving a 31% improvement in docking success rates and up to a 300% improvement when considering the physical validity of the predictions.
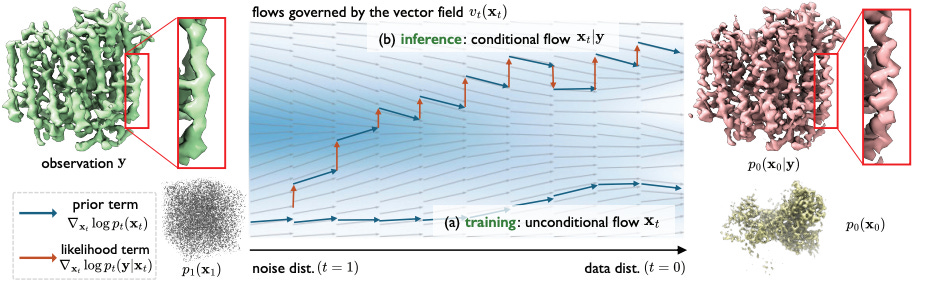
CryoFM: A Flow-based Foundation Model for Cryo-EM Densities
Cryo-electron microscopy (cryo-EM) is an important tool in structural biology and drug discovery that allows scientists to study biomolecules in high detail, with over 38,626 protein density maps produced thanks to advancements in the field. However, the algorithms used to process cryo-EM data haven’t fully utilized the wealth of available biomolecular density maps. To address this, researchers developed CRYOFM, a generative foundation model that learns the distribution of high-quality density maps and can generalize to various downstream tasks. Built on flow matching, CRYOFM effectively captures the prior distribution of these biomolecular density maps. They also introduced a flow posterior sampling method that allows CRYOFM to serve as a flexible prior for multiple downstream tasks in cryo-EM and cryo-electron tomography (cryo-ET) without needing fine-tuning.
ML for Omics
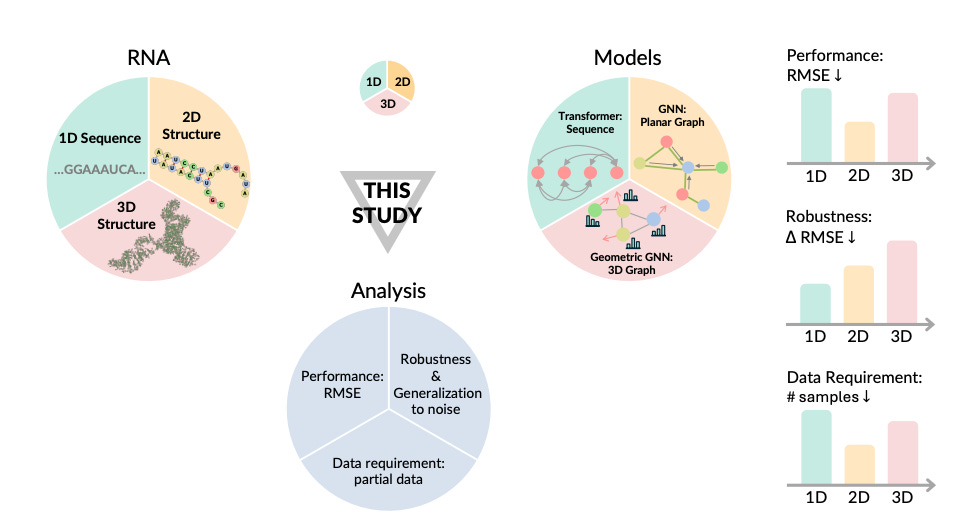
Beyond Sequence: Impact of Geometric Context for RNA Property Prediction
Predicting RNA properties like stability and interactions is key to better understanding biological processes and developing RNA-based therapies. RNA can be represented in different ways—like 1D sequences, 2D graphs, or 3D models—which offer varying levels of insight. Most existing models focus on the 1D sequence but miss out on the geometric context of 2D and 3D structures. This study is the first to systematically evaluate the benefits of incorporating 2D and 3D geometry into RNA property predictions. It shows that models using this geometric data tend to outperform sequence-based models, especially when data is limited or incomplete. However, sequence-based models are more resilient to noisy data, though they need significantly more training data to match the performance of geometry-aware models.
Reviews
Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
LoGG
Derivative-Free Guidance in Continuous and Discrete Diffusion Models with Soft Value-Based Decoding by Xiner Li and Masatoshi Uehara.
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋