


Portal Weekly #68: RNA-LLMs, LLM hallucinations, walk-jump sampling, constant-pH metadynamics simulations, and more.
Join Portal and stay up to date on research happening at the intersection of tech x bio 🧬


Hi everyone 👋
Welcome to another issue of the Portal newsletter where we provide weekly updates on talks, events, and research in the TechBio space!
🚨 Resource Alert
Over the past few years, we’ve seen companies release various open-source datasets to the community via competitions (i.e. Recursion’s rxrx.ai phenomics datasets). These are often great resources for the community. That’s why a few months ago, we were excited to see Leash Bio release BELKA, a massive molecule-protein interaction dataset with some 4.25B physical measurements via a Kaggle competition.
The full dataset is now available through Polaris and can be downloaded with just a few lines of code. If you didn’t get to participate in the competition or just want to explore the dataset in its entirety, now’s your chance!
💬 Upcoming talks
LoGG continues next week with a talk by Giannis Daras Pacesa to discuss training ambient diffusion models with noisy data. Please note that LoGG now starts at 12 pm ET on Mondays!

Join us live on Zoom on Monday at 12 pm ET. Find more details here.
If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
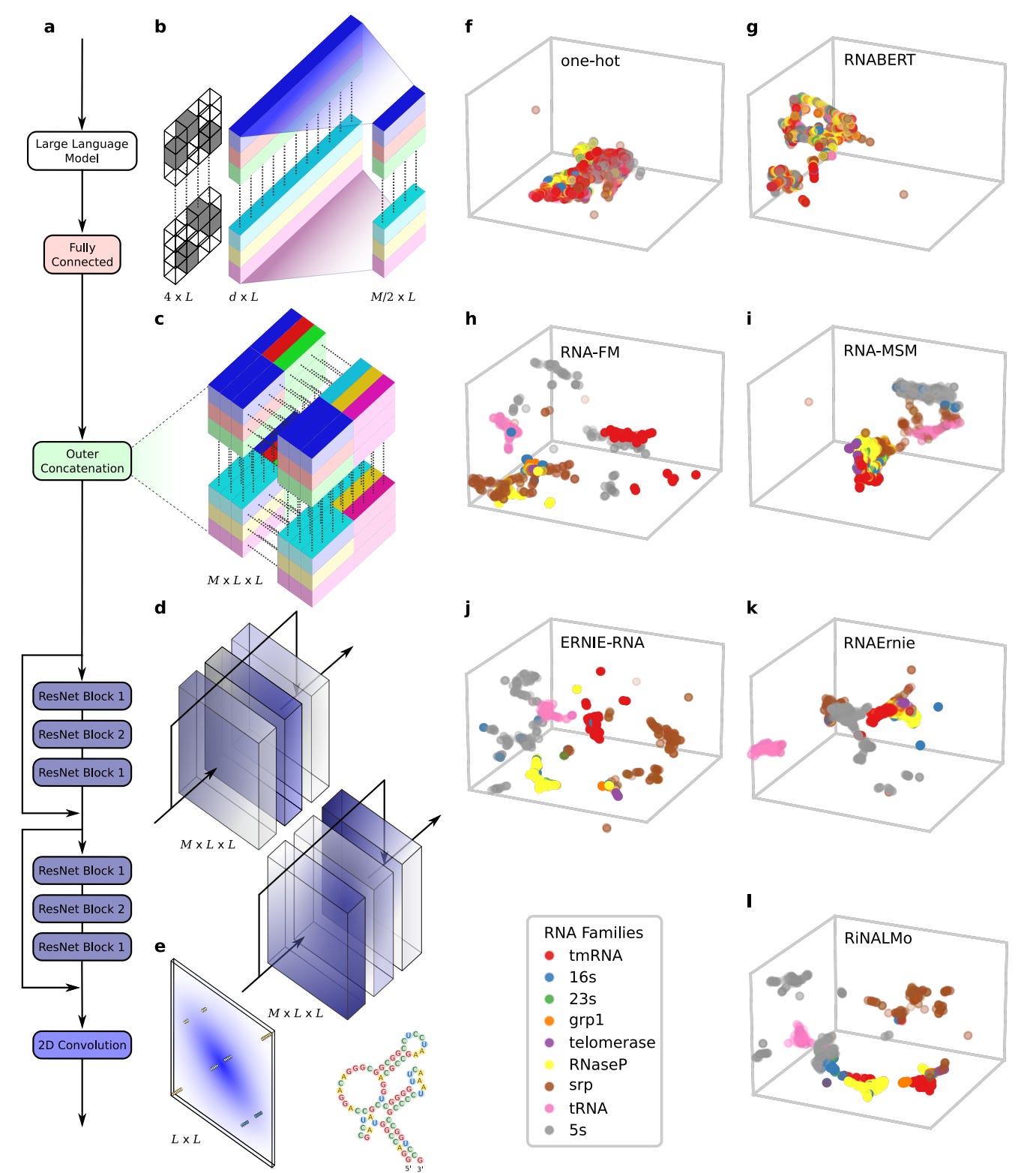
Comprehensive benchmarking of large language models for RNA secondary structure prediction
Following the success of large language models (LLMs) for DNA and proteins, recent efforts have focused on developing LLMs specifically for RNA. These RNA-LLMs leverage extensive datasets of RNA sequences to learn semantically rich numerical representations of each RNA base through self-supervised learning. In this study, researchers conducted a thorough experimental analysis of various pre-trained RNA-LLMs, assessing their performance in predicting RNA secondary structures within a unified deep-learning framework. Results indicated that two LLMs significantly outperformed the others, highlighting notable challenges in generalization when faced with low-homology scenarios.
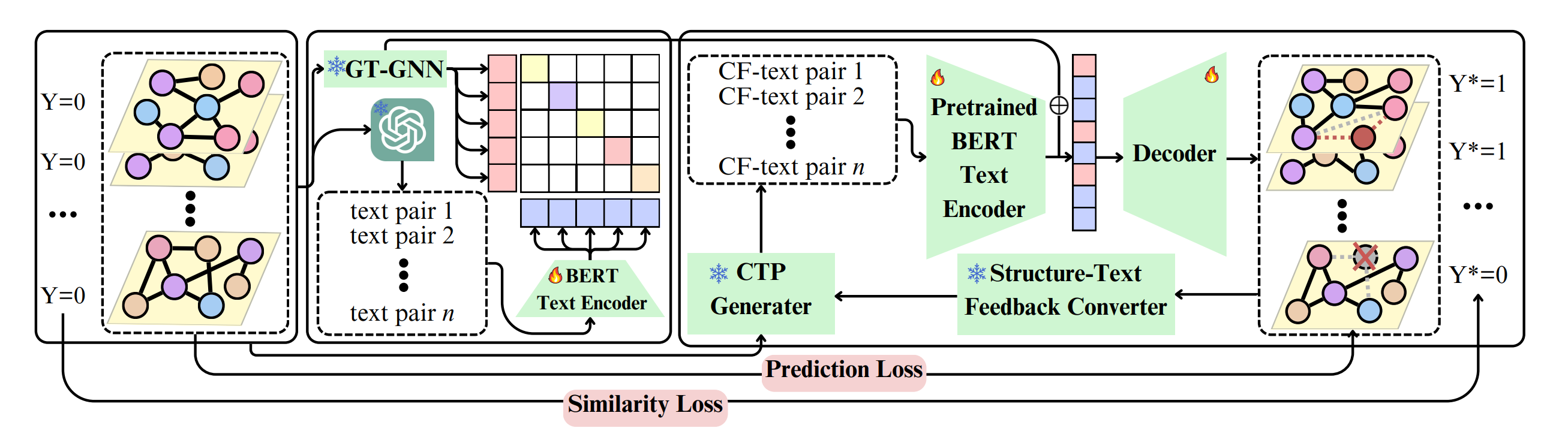
Explaining Graph Neural Networks with Large Language Models: A Counterfactual Perspective for Molecular Property Prediction
In recent years, Graph Neural Networks (GNNs) have shown significant success in molecular property prediction tasks, such as toxicity analysis. However, the black-box nature of GNNs raises concerns when used in high-stakes decision-making scenarios like drug discovery. Researchers propose a novel GCE method called LLM-GCE, which harnesses the capabilities of large language models (LLMs) to explain GNNs in the context of molecular property prediction. The approach uses an autoencoder to generate counterfactual graph topologies from a set of counterfactual text pairs (CTPs) derived from the input graph. Additionally, a CTP dynamic feedback module is integrated to reduce LLM hallucinations, providing intermediate feedback from the generated counterfactuals to enhance guidance.
ML for Small Molecules
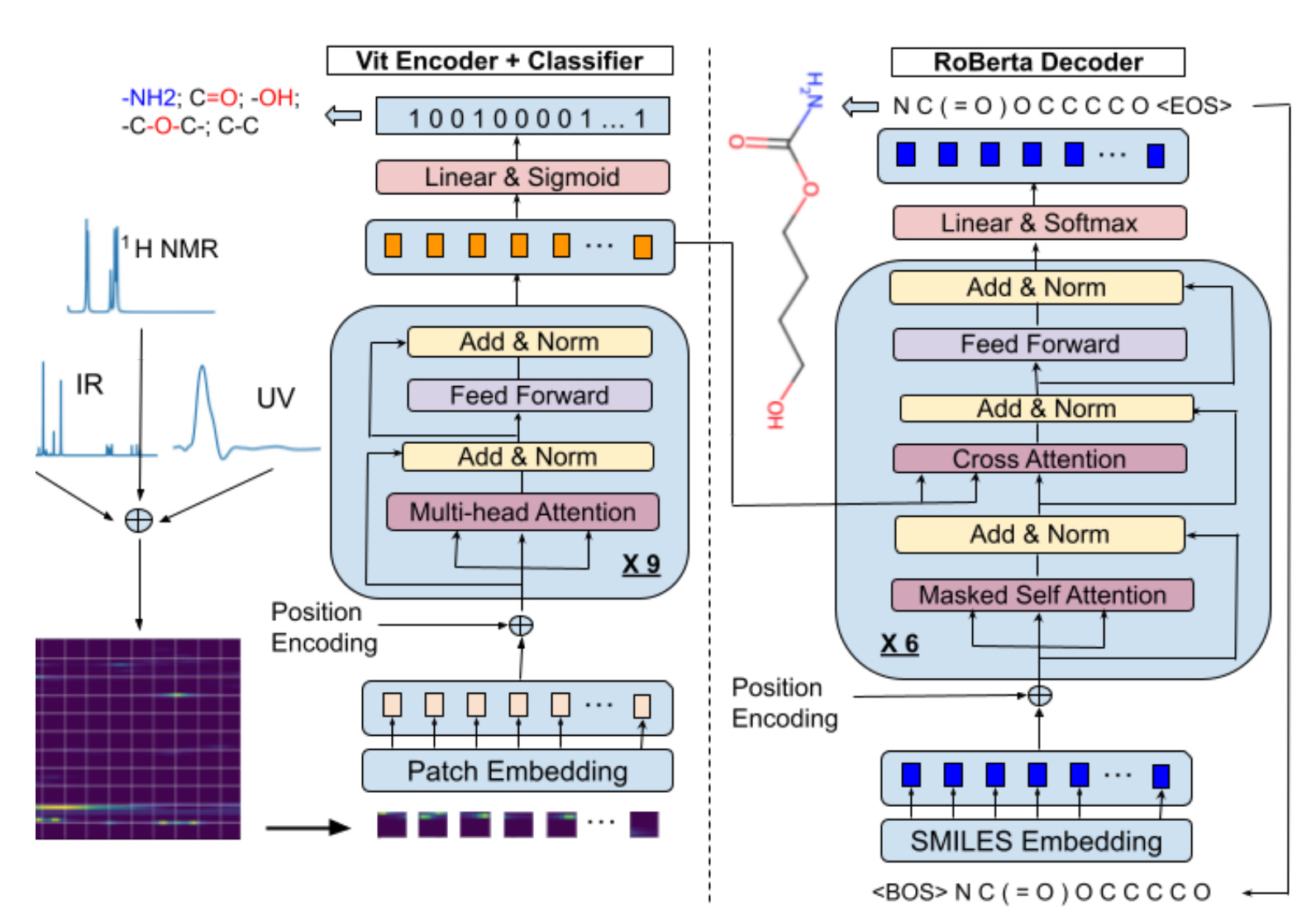
A Transformer Based Generative Chemical Language AI Model for Structural Elucidation of Organic Compounds
For over 50 years, computer-aided structural elucidation systems (CASE) for organic compounds have depended on complex expert systems with explicitly programmed algorithms. These systems can be computationally inefficient when dealing with complex compounds due to the extensive chemical structural space that needs to be explored and filtered. In this study, researchers introduce a transformer-based generative chemical language AI model, an innovative end-to-end architecture aimed at replacing the traditional logic and workflow of classic CASE systems for rapid and accurate spectroscopic-based structural elucidation. The model utilizes an encoder-decoder architecture along with self-attention mechanisms similar to those found in large language models, enabling it to directly generate the most probable chemical structures that correspond to the input spectroscopic data.
ML for Atomistic Simulations
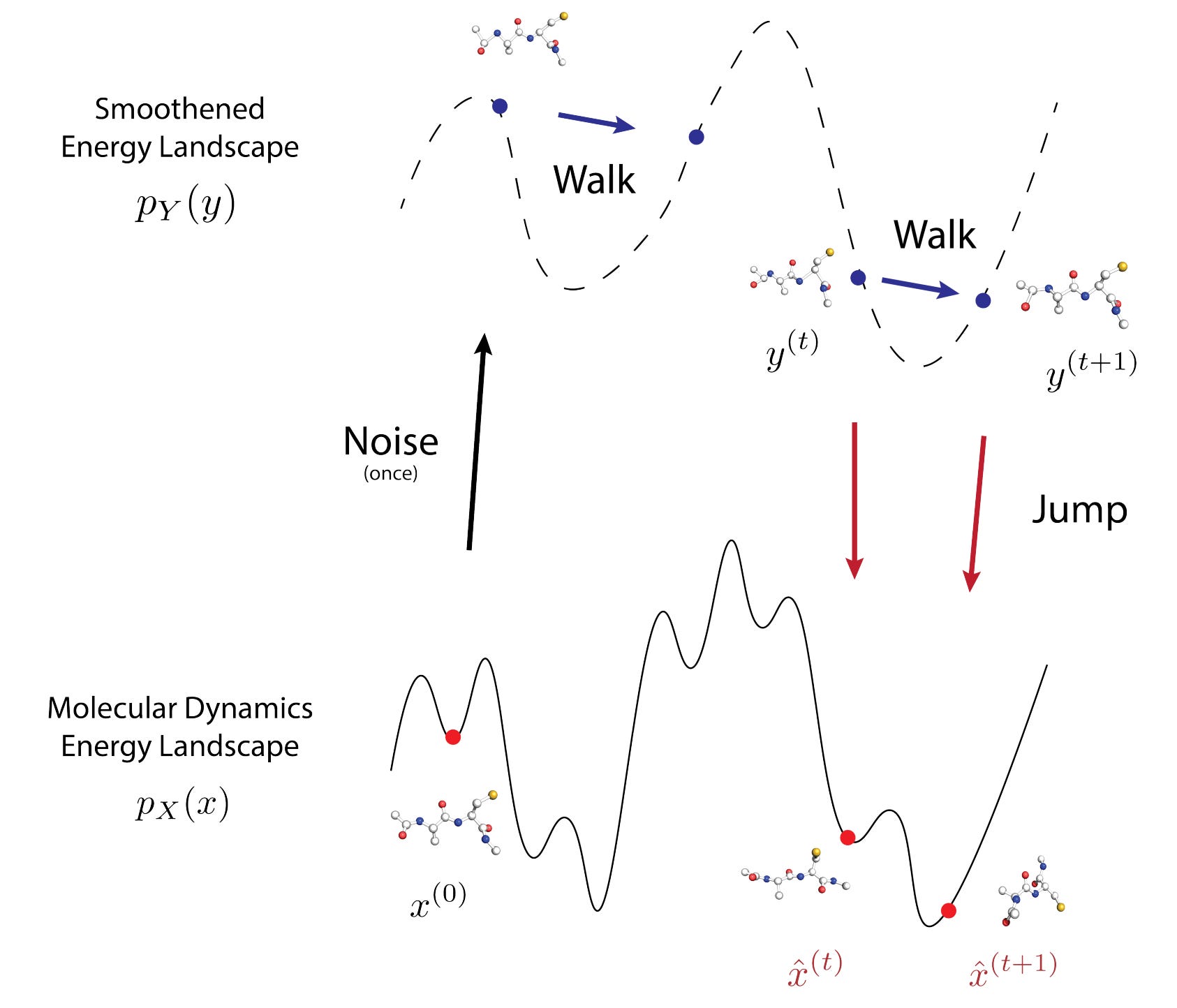
JAMUN: Transferable Molecular Conformational Ensemble Generation with Walk-Jump Sampling
Understanding protein function and advancing drug discovery, especially for innovative approaches like targeting cryptic pockets, hinges on the study of conformational ensembles of protein structures. However, current methods for sampling these ensembles often struggle with computational efficiency and lack adaptability to systems beyond their training data. Enter walk-Jump Accelerated Molecular ensembles with Universal Noise (JAMUN). This new approach marks a significant step towards efficiently sampling the Boltzmann distribution of various proteins. By extending Walk-Jump Sampling techniques to point clouds, JAMUN can generate ensembles at speeds that far surpass traditional molecular dynamics and even cutting-edge machine learning methods.
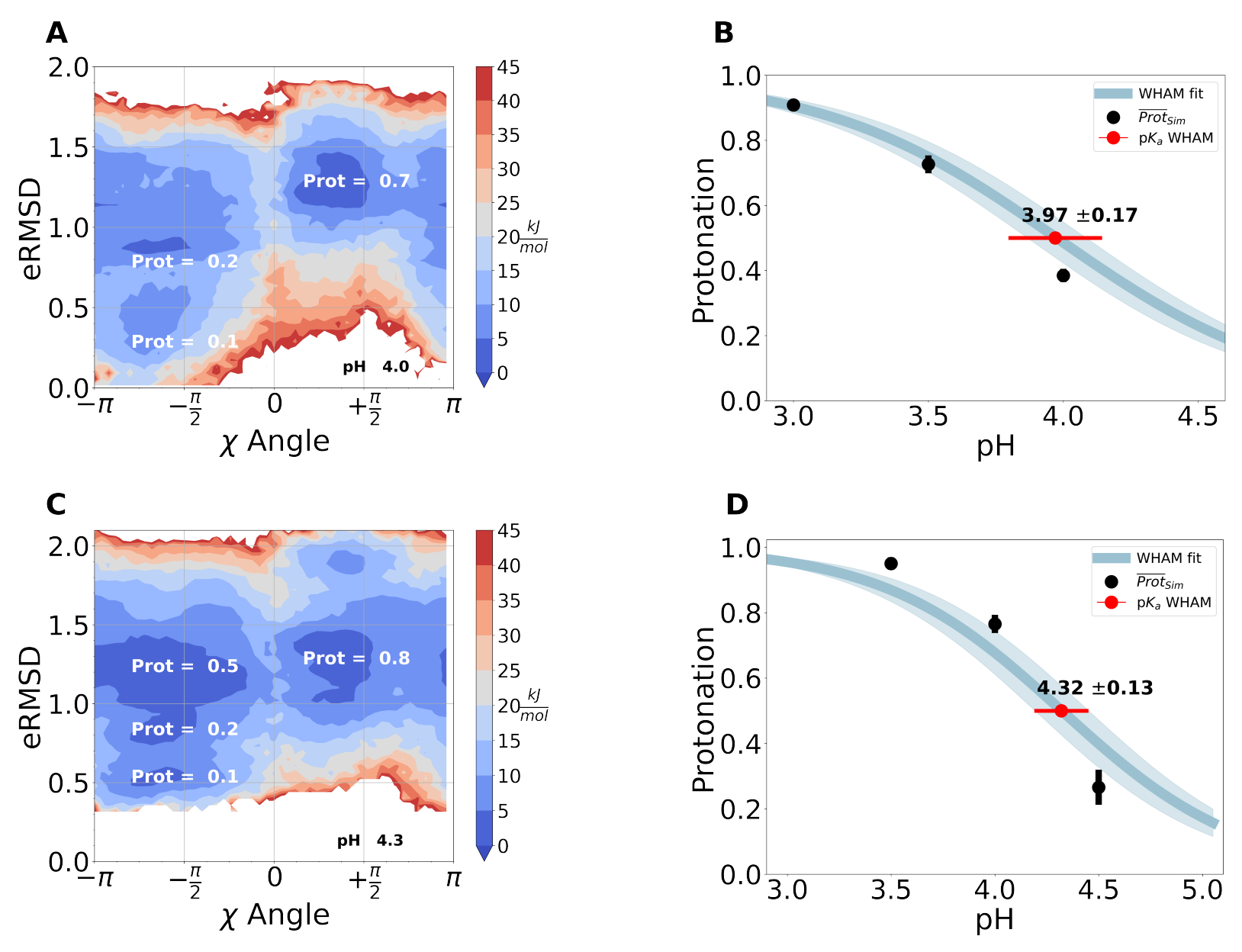
Characterizing RNA oligomers using Stochastic Titration Constant-pH Metadynamics simulations
RNA molecules are vital for a range of biological functions, largely due to their diverse and flexible structures. This flexibility is driven by complex hydrogen-bonding networks formed by both canonical and non-canonical base pairs, which are influenced by protonation events that stabilize or disrupt these interactions. In this study, researchers extended the stochastic titration CpHMD method to incorporate RNA parameters from the standard χOL3 AMBER force field. They demonstrated its effectiveness in capturing titration events of nucleotides in single-stranded RNAs. Validation was performed using trimers and pentamers with a single central titratable site, integrating a well-tempered metadynamics approach (CpHMetaD) via PLUMED.
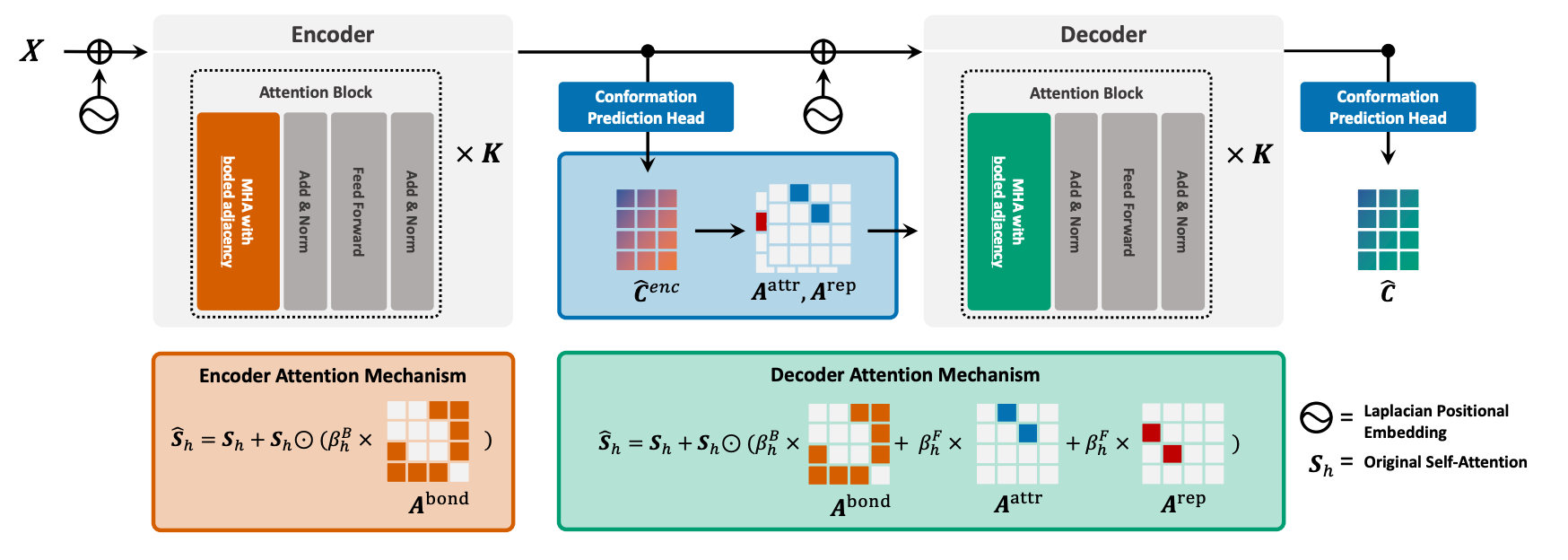
Rebind: Enhancing Ground-State Molecular Conformation Prediction via Force-Based Graph Rewiring
Recently, deep learning (DL) techniques have emerged as promising alternatives to computationally intensive classical methods like density functional theory (DFT). However, existing DL approaches often fall short in accurately modeling inter-atomic forces, especially for non-bonded atomic pairs, due to their simplistic reliance on bonds and pairwise distances. To tackle this issue, researchers have developed REBIND, a novel framework that rewires molecular graphs by introducing edges based on the Lennard-Jones potential. This adjustment helps capture non-bonded interactions for low-degree atoms more effectively. Experimental results indicate that REBIND significantly outperforms state-of-the-art methods across a range of molecular sizes, achieving up to a 20% reduction in prediction error.
ML for Proteins
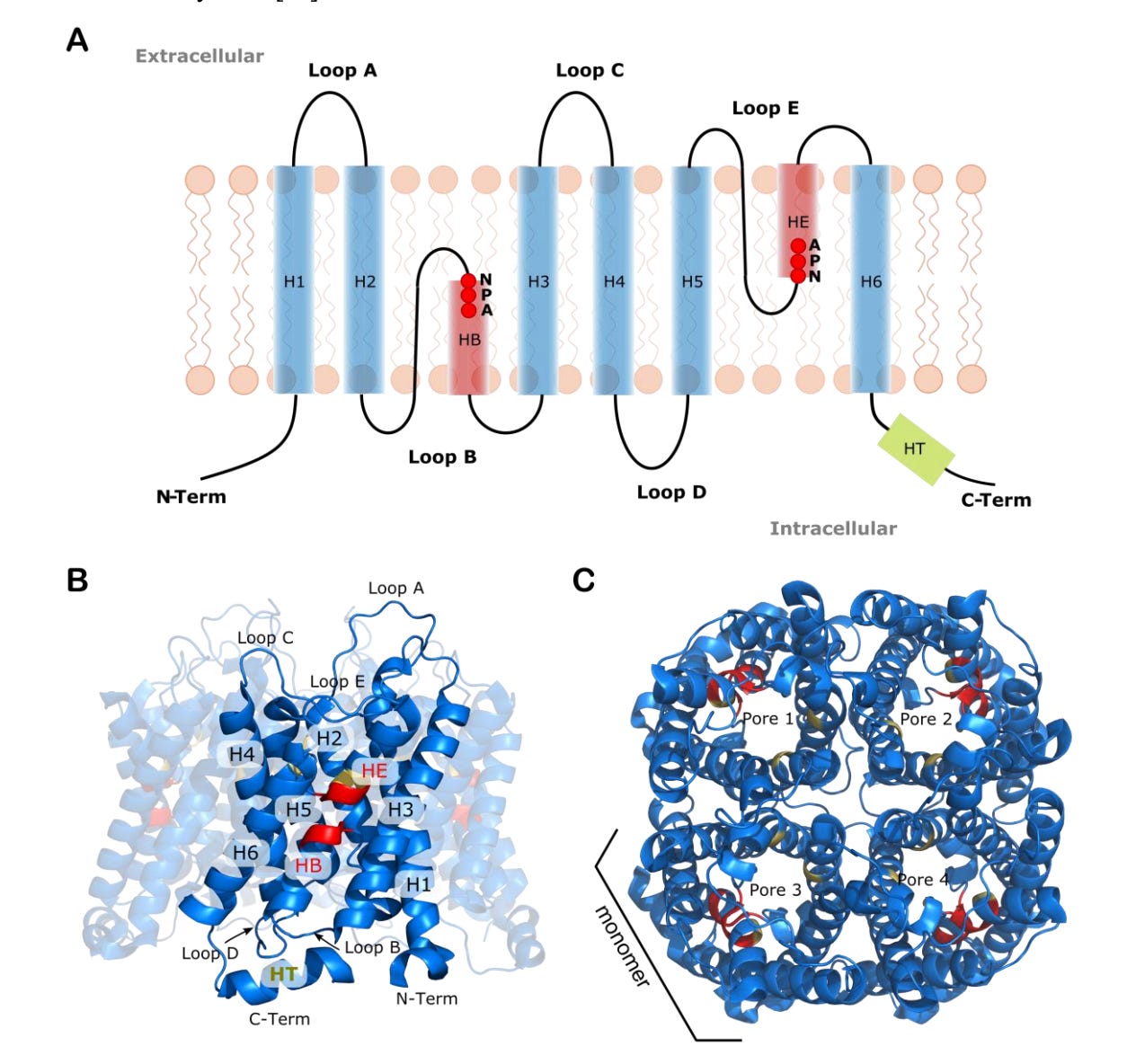
Functionality Determinants and Pore-level Quantitative Structure–Activity Relationship (QSAR) Approach for Water Permeation Rate in Aquaporins
Aquaporins (AQPs) are crucial for regulating water transport and solute selectivity in biological cells, leading to growing interest in their application for biomimetic membranes in water filtration. This study conducts a thorough analysis of AQPs' structural features to enhance the design of biomimetic molecules with targeted filtration capabilities. Focusing on a selection of experimentally resolved AQP structures, the researchers examine the NPA and Selectivity Filter motifs, structural similarities, pore shapes, and residue-level lining. They then correlate these structural and physicochemical descriptors with experimental permeation rates using pore-level QSAR modeling.
ML for Omics
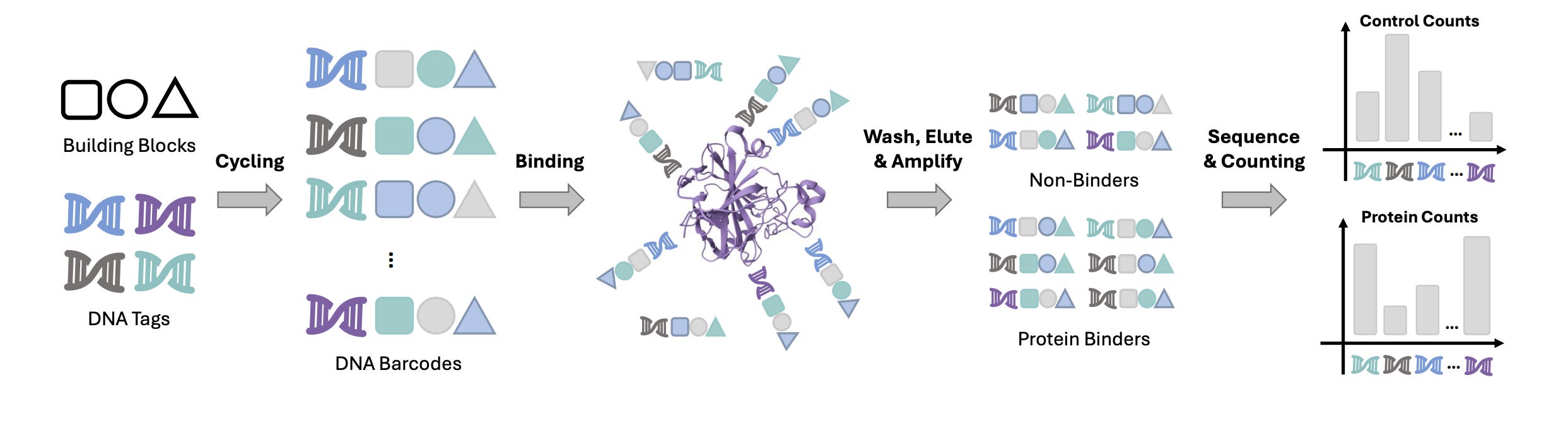
Del-Ranking: Ranking-Correction Denoising Framework for Elucidating Molecular Affinities in DNA-Encoded Libraries
DNA-encoded library (DEL) screening has significantly changed how protein-ligand interactions are detected using read counts, allowing researchers to rapidly explore vast chemical spaces. However, noise in these read counts, often due to nonspecific interactions, can distort this exploration. To tackle these challenges, researchers have developed DEL-Ranking, a novel distribution-correction denoising framework. This approach introduces two main innovations: first, a new ranking loss that adjusts the relative magnitude relationships between read counts, helping the model learn causal features that determine activity levels; second, an iterative algorithm that uses self-training and consistency loss to ensure coherence between activity label predictions and read counts. Extensive evaluations across various DEL datasets have shown that DEL-Ranking outperforms existing methods on multiple correlation metrics, significantly improving binding affinity prediction accuracy.
Reviews
Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
LoGG
BindCraft: one-shot design of functional protein binders by Martin Pacesa
Meta Flow Matching: Integrating Vector Fields on the Wasserstein Manifold by Lazar Atanackovic.
You can always catch up on previous recordings on our YouTube channel or the Portal Events page!
Portal is the home of the TechBio community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
CARE - a weekly reading group to discuss the latest research in causality
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋