


📫 Community Round-Up: Week of October 31st, 2023
A new blog post on designing functional molecules with desired properties! Also: AlphaFold bulks up, a knowledge graph foundation model, an executive order, and many more papers.

Hi everyone 👋
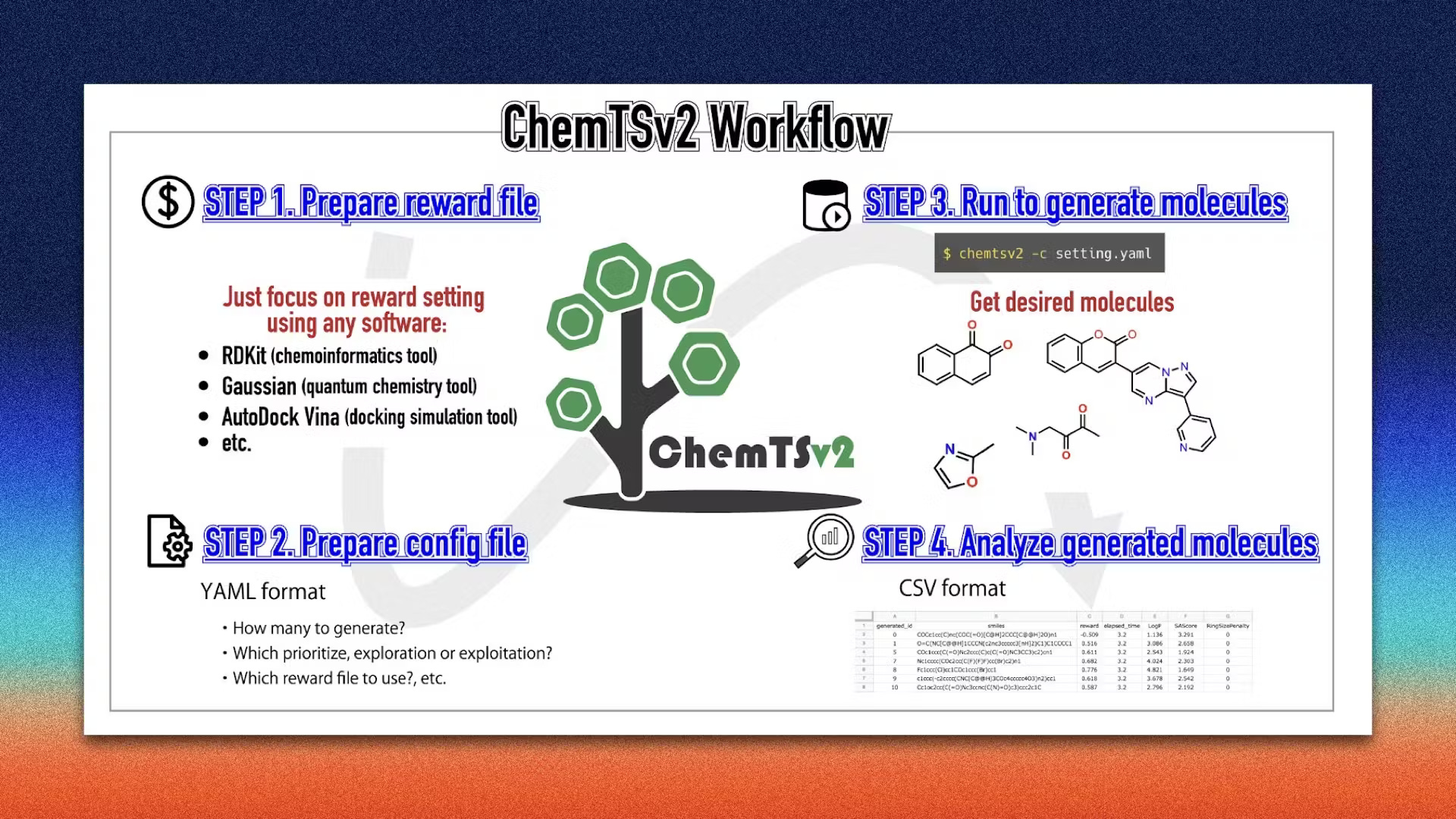
Welcome to another issue of the community newsletter! We just released a new community blog post written by Shoichi Ishida from Yokohama City University, who discusses ChemTSv2, an open-source AI-based tool for designing functional molecules with desired properties. Check it out!
M2D2 Talks continue next week with a presentation by Eric Nguyen from Stanford who will present HyenaDNA, modelling long-range genomic sequences at single-nucleotide resolution.
Join us live on Zoom on Tuesday, November 7th at 11 am ET. Find more details here.

The Molecular Machine Learning (MoML) conference is next week! The conference will be hosted at MIT on Wednesday, November 8th 2023. Grab your tickets or sign up for the waitlist here.

If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
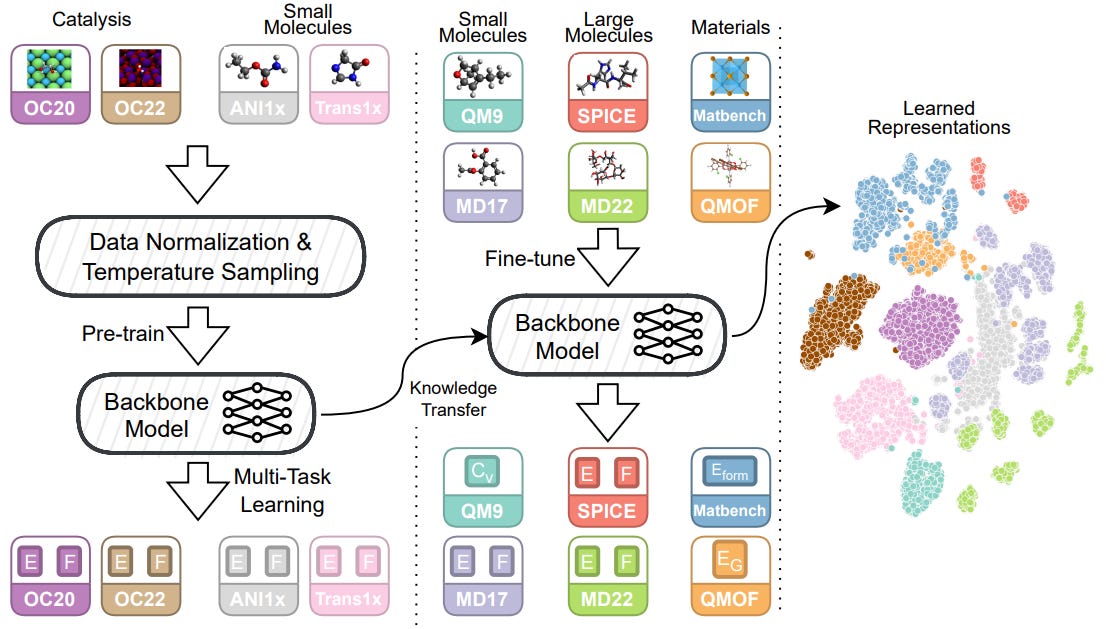
From Molecules to Materials: Pre-training Large Generalizable Models for Atomic Property Prediction
Foundation models (large generalizable models with transferability to downstream tasks) are sought in many sub-disciplines in science. This study introduces Joint Multi-domain Pre-training (JMP), a method for training models to predict atomic properties across different chemical domains. JMP pre-trains models using multiple datasets from various chemical domains and then fine-tunes them on downstream tasks. The results show that JMP outperforms training models from scratch, achieving an average improvement of 59% and matching or setting state-of-the-art results on 34 out of 40 tasks, emphasizing the potential of diverse data pre-training for improving property prediction in different chemical domains, particularly for low-data tasks.
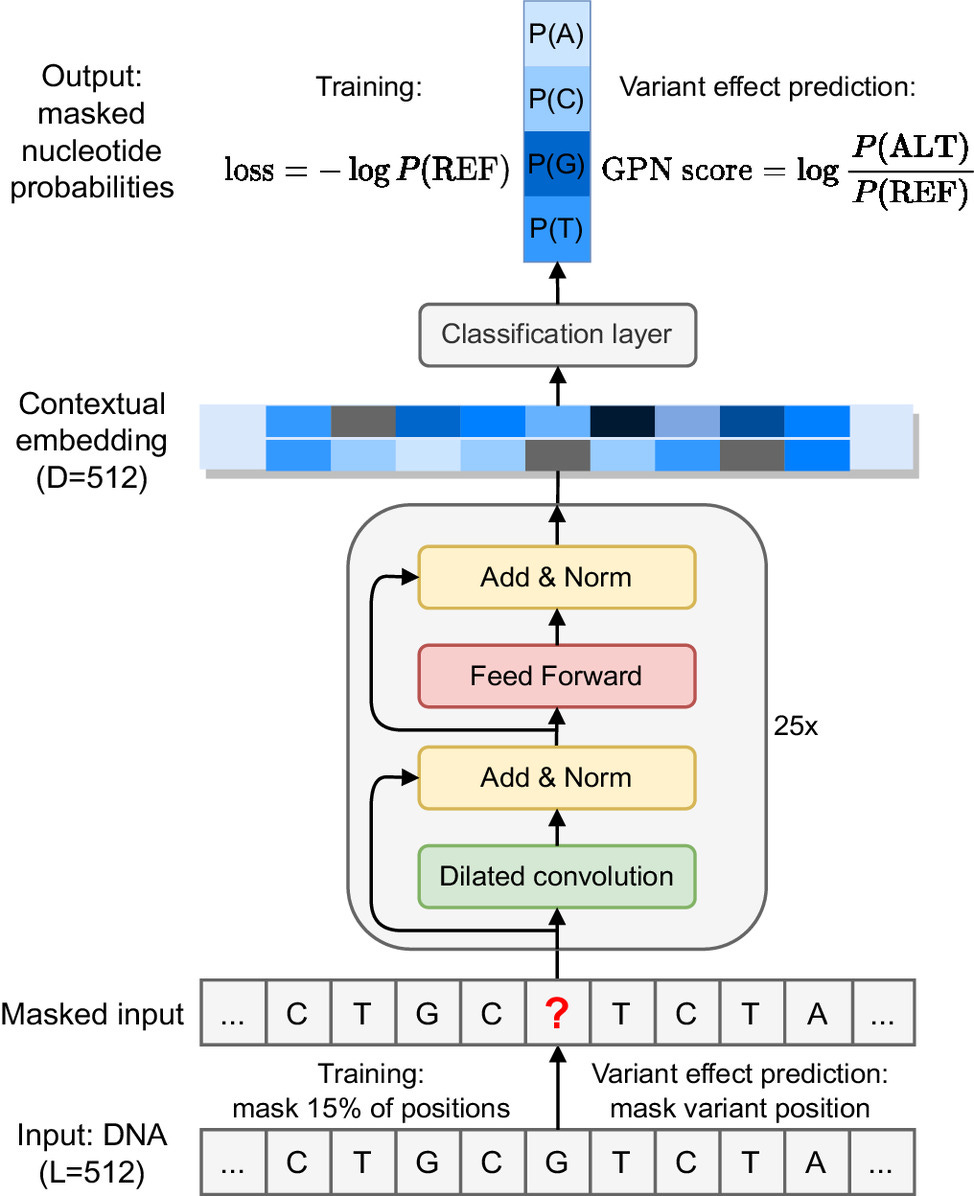
DNA Language Models are Powerful Predictors of Genome-Wide Variant Effects
Genetic variants across the genome affect a range of phenotypic outcomes but biological complexity makes it difficult to interpret which variant has what effect. This study uses an unsupervised DNA language model to predict genetic variants in the plant model organism Arabidopsis thaliana, outperforming popular conservation-score-based approaches. The model learns gene structure and DNA motifs without supervision, so it can be applied any species with a reference genome.
Graph Learning
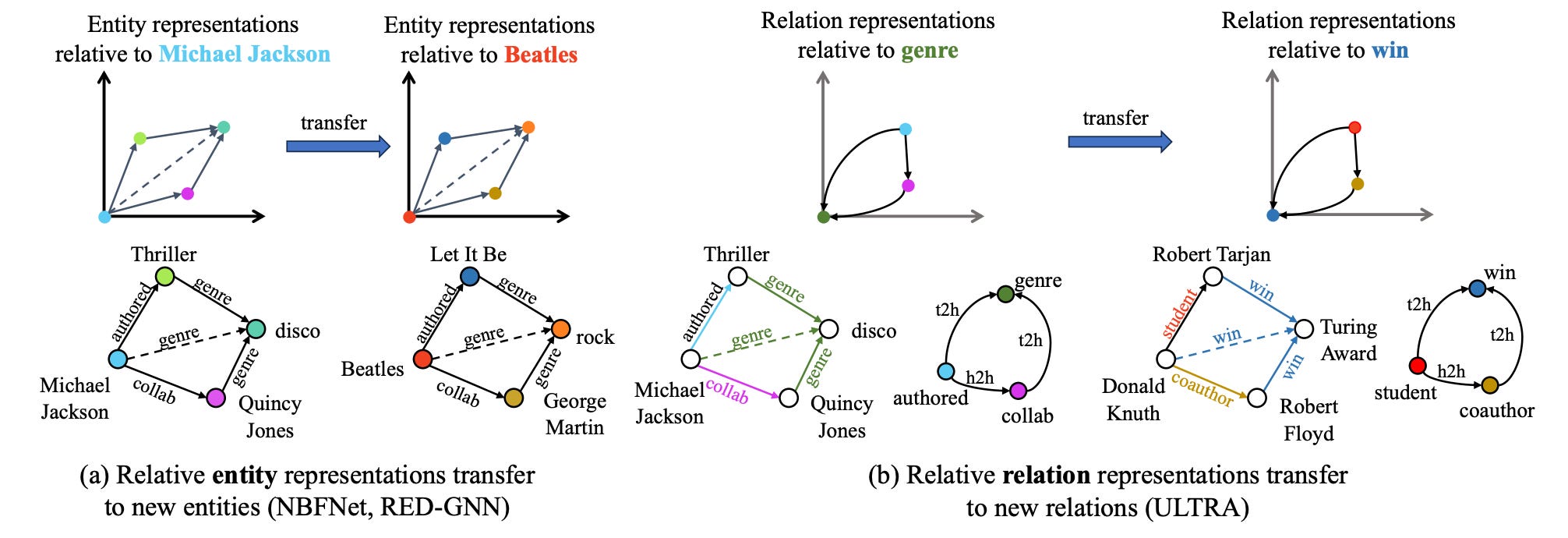
Towards Foundation Models for Knowledge Graph Reasoning
Developing foundation models for knowledge graphs (KGs) is challenging because they have different entity and relation vocabularies that generally don’t overlap. The authors introduce ULTRA, an approach for learning universal and transferable graph representations that can perform inference on any KG with different vocabularies. They show that a pre-trained ULTRA model can generalize well to unseen KGs in zero-shot inductive inference experiments, often outperforming models trained specifically for individual graphs, and fine-tuning further improves its performance.
ML for Small Molecules
Neural Scaling of Deep Chemical Models
In many areas of ML, bigger models seem to do better — computer vision and natural language processing have benefited hugely from training bigger and bigger models. Scale may also be important in scientific deep learning, but incorporating physical priors in chemical deep learning can somewhat compensate if you don’t have huge datasets. This study develops strategies for scaling deep chemical models and looks at how performance scales over dataset and model sizes. They introduce a ChemGPT model with up to 1 billion parameters trained on up to 10 million molecules from PubChem. Ultimately, they find that model loss keeps decreasing with larger models and data sizes, and identify power-law scaling relations.

Searching for High-Value Molecules Using Reinforcement Learning and Transformers
Reinforcement learning (RL) can be useful for finding high-value policies. This study conducts >100 experiments with different RL algorithms, pre-training methods, and molecular grammars to search the vast space of molecular graphs and explore how different design choices for text grammar and algorithmic choices for training can affect an RL policy's ability to generate molecules with desired properties. Their final RL-based molecular design algorithm ends up being more straightforward than previous work, and shows which design choices are actually helpful for text-based molecule design.
ML for Atomistic Simulations
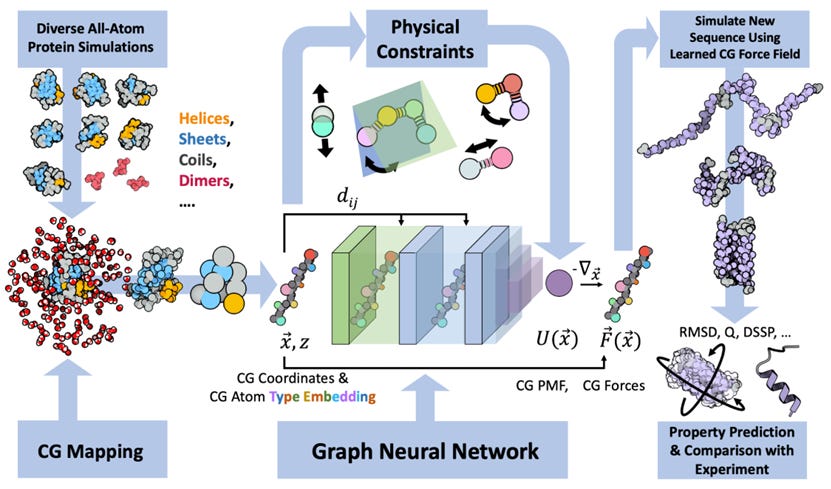
Navigating Protein Landscapes With a Machine-Learned Transferable Coarse-Grained Model
All-atom molecular dynamics are powerful for protein simulations but costly. The search for a machine-learned model for proteins, which would be a lot faster, is an ongoing challenge. This study develops a coarse-grained force field that can be used for extrapolative molecular dynamics on new sequences not used during model parameterization. The model successfully predicts folded structures, intermediates, metastable folded and unfolded basins, and the fluctuations of intrinsically disordered proteins while being several orders of magnitude faster than an all-atom model.
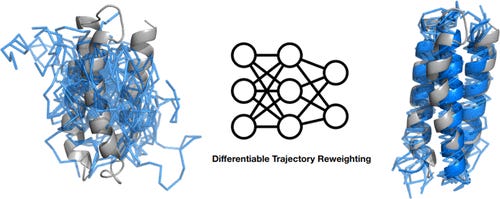
Top-Down Machine Learning of Coarse-Grained Protein Force Fields
A different study working to develop coarse-grained representations of proteins. They simulate proteins using molecular dynamics (MD), then use the trajectories to train a neural network potential through differentiable trajectory reweighting. They say that this only needs native conformations of proteins (experimental static structures), and the model can extrapolate to cases outside of the training distribution.
ML for Proteins
A Glimpse of the Next Generation of AlphaFold
Google Deepmind and Isomorphic Labs recently shared an update on the latest progress of the AlphaFold protein structure prediction model. They don’t present any code but do show a few graphs suggesting improvements of the latest AlphaFold version over AlphaFold2. The model now does better on small molecules, proteins, nucleic acids (DNA and RNA) and post-translational modifications, bringing it in line with RoseTTAFold All-Atom, which we featured a few weeks ago. Can’t wait to see a more direct comparison between the two once they become available!

Accurate and Fast Prediction of Intrinsically Disordered Protein by Multiple Protein Language Models and Ensemble Learning
Intrinsically disordered proteins (IDPs) aren't well modeled by the current crop of structure modelling techniques because they don't crystallize well so they don't appear in datasets. ML methods that have had success in predicting IDPs focus on the amino acid sequence and build up from there. This study uses a representation extracted from several SOTA protein language model and predicts intrinsically disordered regions by ensemble learning with bidirectional recurrent neural nets. This kind of a tool can be fairly accurate and fast for proteome-level analysis, helping researchers focus on particular proteins (or avoid them, if you don't want intrinsically disordered regions).

ML for Omics
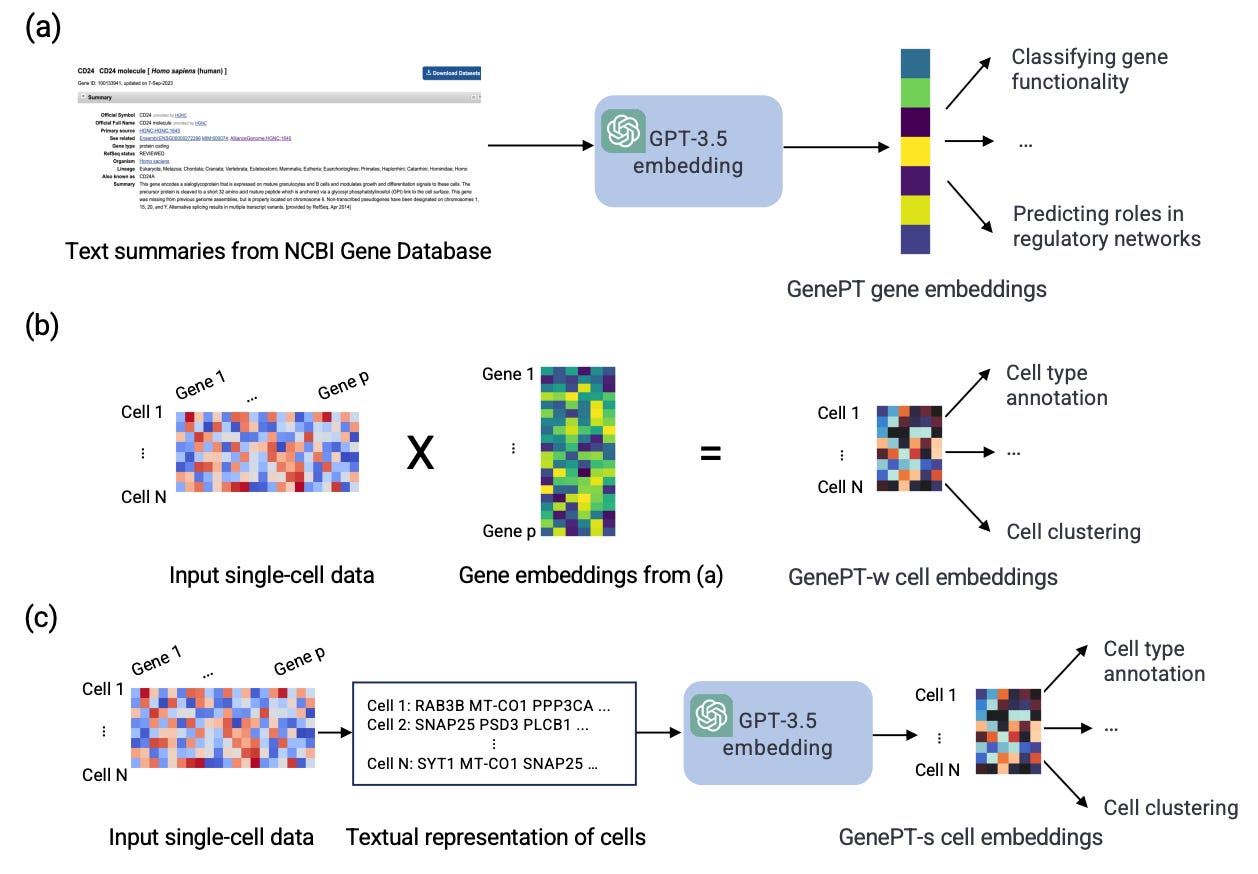
GenePT: a Simple but Hard-to-Beat Foundation Model for Genes and Cells Built from ChatGPT
Most foundation models for single-cell transcriptomes use large-scale gene expression data. This study instead explores leveraging ChatGPT embeddings of genes based on literature. GenePT uses NCBI text descriptions of individual genes with GPT-3.5 to generate gene embeddings. GenePT performs comparably or even better than other recent single-cell foundation models on various downstream tasks like classifying gene properties and cell types. This approach is worth exploration, since it is efficient and easy to use, and doesn’t need dataset curation and additional pre-training.
Causal Machine Learning for Single-Cell Genomics
Advancements in single-cell omics and large-scale perturbation screens offer opportunities to study gene causality in complex biological processes, but challenges arise due to high-dimensional data and biological complexity. The machine learning community is increasingly interested in applying causal techniques to single-cell genomics. This perspective outlines the approach, challenges in generalization and interpretability, and suggests research directions. Causal models are expected to become vital tools for informed experimental design with the growth of single-cell data.

Open Source
ORDerly: Datasets and Benchmarks for Chemical Reaction Data
Chemical reactions are the backbone of experimental life sciences. lack of high-quality open-source chemical reaction datasets for training ML models. This paper presents an open-source Python package for customizable and reproducible preparation of reaction data stored in accordance with the increasingly popular Open Reaction Database (ORD) schema. This resource will be useful for cleaning and preparing large chemical reaction data.
Other News
White House Executive Order on the Safe, Secure, and Trustworthy Development and Use of Artificial Intelligence
AI safety in general is very much in the public consciousness, and the United States government is responding. Among the main takeaways for those of us interested in AI for drug discovery are that some (very large) models and computing clusters will require reporting going forward. Luckily (for most of us), the requirement is for “any model that was trained using a quantity of computing power greater than 1026 integer or floating-point operations, or using primarily biological sequence data and using a quantity of computing power greater than 1023 integer or floating-point operations”, which currently affects only the big players in the space.
Think we missed something? Join our community to discuss these topics further!
🗃️ Latest Recordings
M2D2
Machine Learning for Multi-Scale Molecular Simulation and Design by Xiang Fu
LoGG
Mirror Diffusion Models for Constrained and Watermarked Generation by Guan-Horng Liu
Valence Portal is the home of the AI for drug discovery community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋