
Portal Weekly #23: TechBio mixers with Michael Bronstein, an upcoming RoseTTAFold All-Atom talk, the impact of LLMs on scientific discovery, active learning to traverse chemical space, and more.
Join Valence Portal and stay up to date on research happening at the intersection of tech x bio 🧬




Hi everyone 👋
Welcome to another issue of the Valence Portal newsletter where we provide weekly updates on everything happening in TechBio research!
📅 Upcoming events
We’ll be hosting two TechBio mixers next week in Oxford and Cambridge!
The Oxford mixer is being co-hosted with Michael Bronstein on November 22nd - RSVP here. The Cambridge event is happening on campus at King’s College on November 23rd - RSVP here. Join the UK TechBio community for an evening of dinner, drinks, and discussions on computational approaches for accelerating drug discovery.
See you next week!
💬 Upcoming talks
M2D2 continues next week with a presentation by Leon Klein and Andrew Foong on their paper Timewarp: Transferable Acceleration of Molecular Dynamics by Learning Time-Coarsened Dynamics.
Join us live on Zoom on Tuesday, November 21st at 11 am ET. Find more details here.

LoGG continues with a presentation by Rohith Krishna from University of Washington who will talk about RoseTTAFold All-Atom, which we featured in a previous newsletter. You won’t want to miss this one!
Join us live on Zoom on Monday, November 20th at 11 am ET. Find more details here.

If you enjoy this newsletter, we’d appreciate it if you could forward it to some of your friends! 🙏
Let’s jump right in!👇
📚 Community Reads
LLMs for Science
The Impact of Large Language Models on Scientific Discovery: a Preliminary Study using GPT-4
Microsoft Research AI4Science and Microsoft Azure Quantum have put out a huge (230 page) report examining the performance of GPT-4, one of the latest large language models (LLMs), on a range of scientific areas including drug discovery, biology, and computational chemistry. Worth taking a more in-depth look!
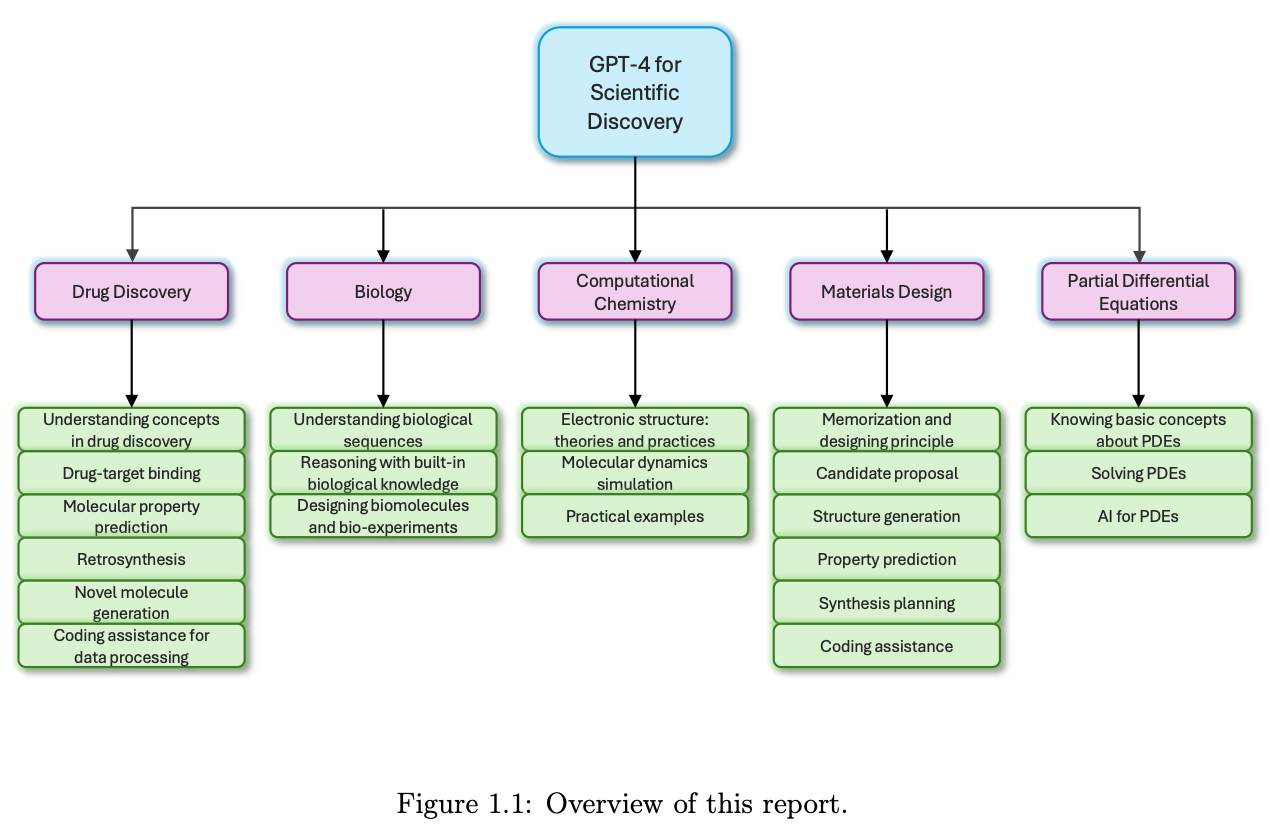
Extracting human interpretable structure-property relationships in chemistry using XAI and large language models
Explainable artificial intelligence (XAI) tries to address the ‘black box’ nature of machine learning models. XAI can be used in chemistry to understand structure-property relationships. This study develops a framework that integrates XAI methods with LLMs to generate accessible natural language explanations of raw chemical data, which could be a useful tool for less technical users.
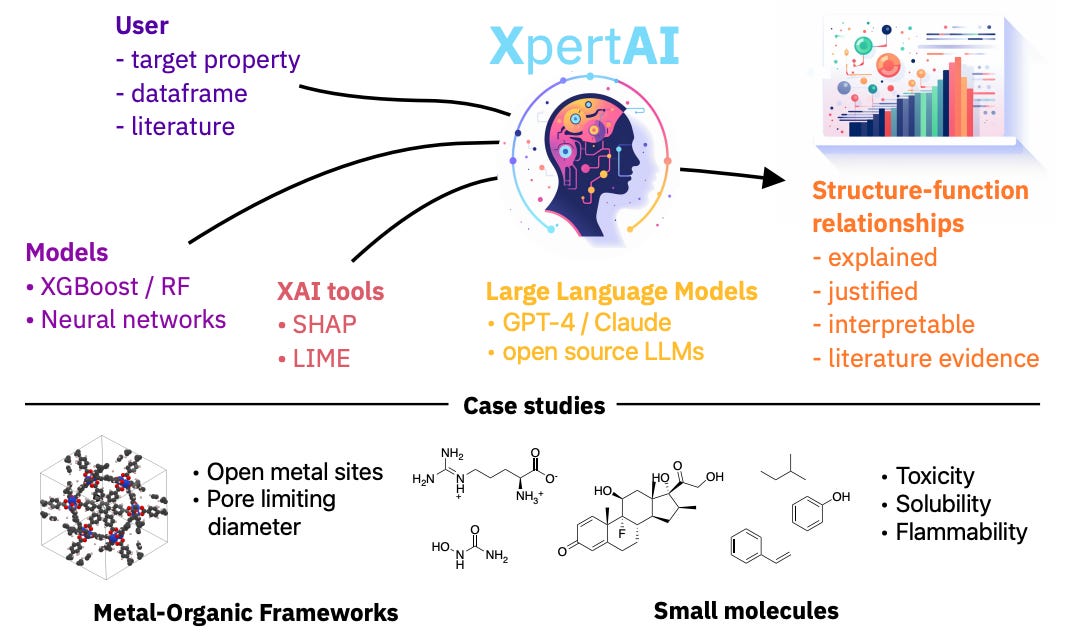
Multi-Modal Learning
CLOOME: contrastive learning unlocks bioimaging databases for queries with chemical structures
Similar to COATI, which we featured in newsletter #16, CLOOME uses contrastive learning, embedding bio images and chemical structures into a common latent space using encoders. This approach allows linking a given chemical structure with an image of a biological phenomenon, and can greatly help understand biological effects of molecules. Their model also shows transferability to other drug discovery tasks, making it a step toward a foundation model for microscopy images.
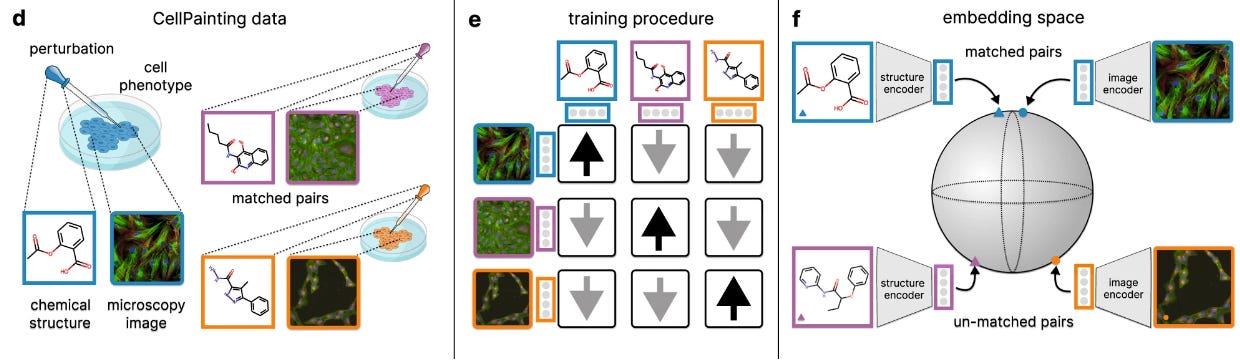
Graph Learning
Calibrated geometric deep learning improves kinase–drug binding predictions
Protein kinases are important: they regulate a range of cellular functions, and kinase inhibitors are one of the largest group of approved drugs. This study presents KDBNet, a model for learning binding affinity between kinases and compounds. By integrating 3D structure information from both kinase binding pocket and molecule into graph neural networks, they get better accuracy in predicting drug-kinase binding affinity compared to 1D or 2D featurizing methods. KDBNet can also exploit uncertainty estimates to guide the learning process.
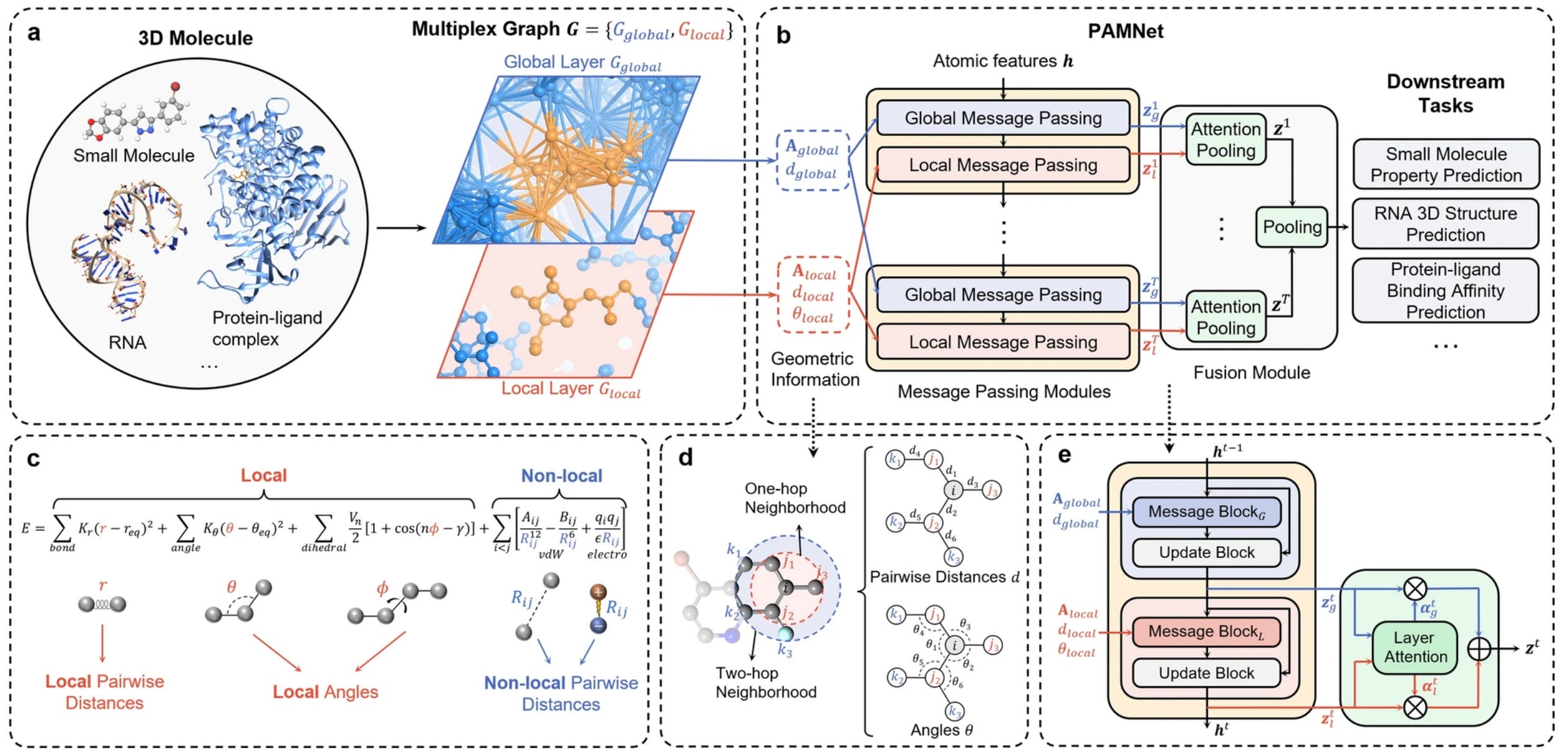
A universal framework for accurate and efficient geometric deep learning of molecular systems
Existing work in molecular science can have limitations for large-scale tasks. This study presents PAMNet as a framework that learns representations of 3D molecules in any molecular system. They use a physics-informed bias to explicitly model interactions and effects, reducing expensive operations. This could be a framework that’s applicable to a range of molecular applications.
ML for Small Molecules
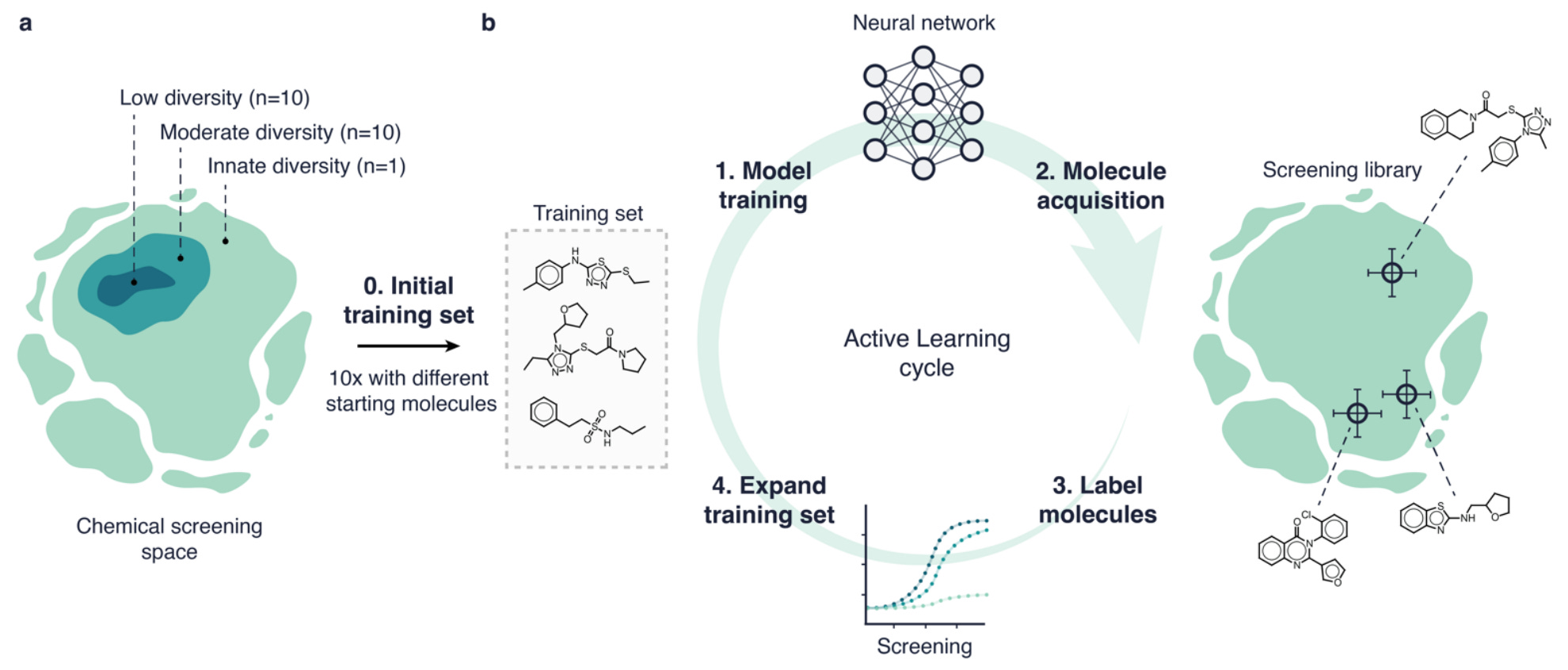
Traversing Chemical Space with Active Deep Learning
Machine learning is limited by the data, and in drug screening there’s often fewer samples and less molecular diversity. Active learning can help by iteratively updating a model by querying a limited set of molecules at a time. This study compared six different active learning strategies and two deep learning models. They found that the strategy to acquire molecules was the primary driver of performance, but that active learning could give up to a six-fold improvement in hit retrieval compared to traditional methods in a low-data scenario.
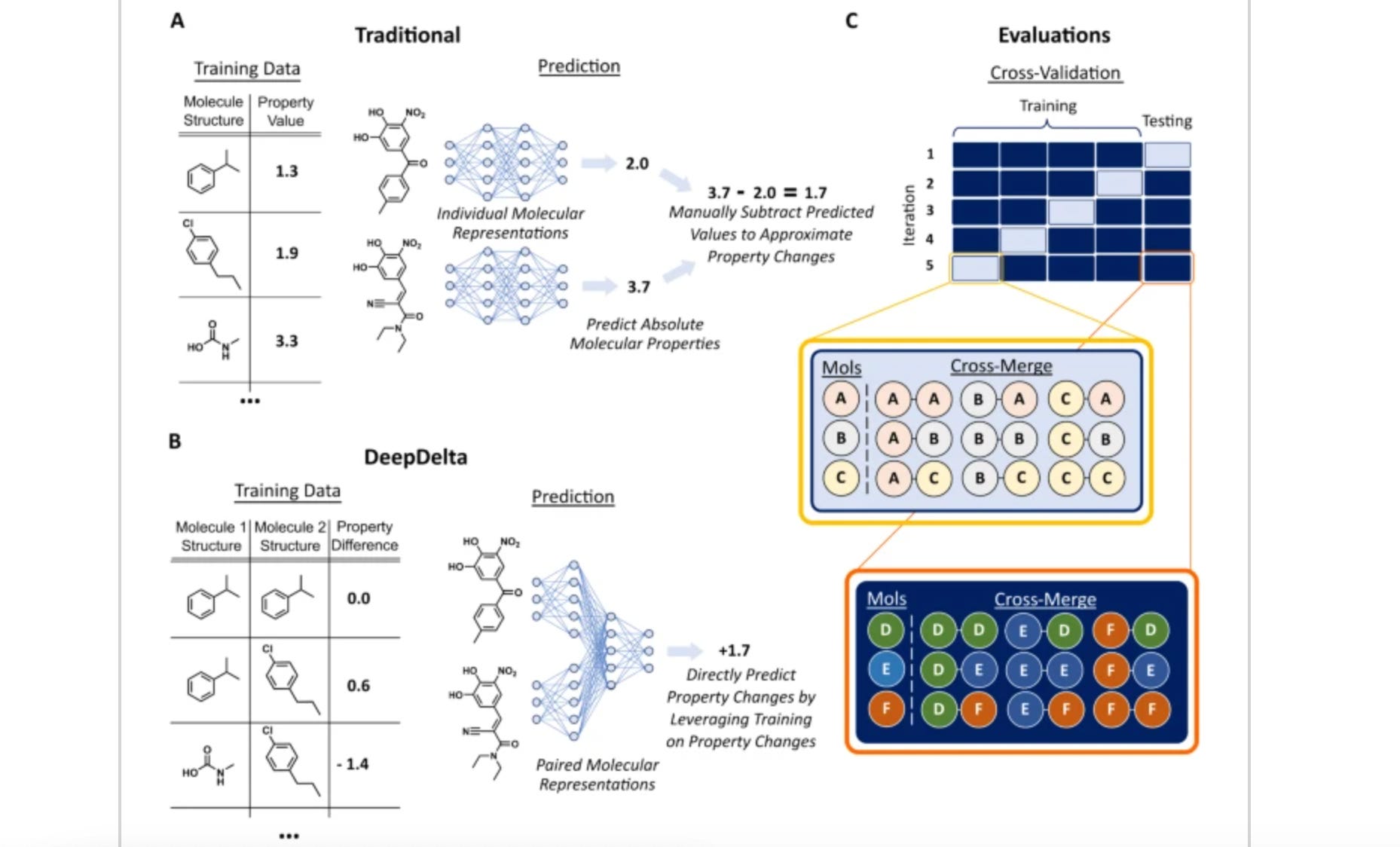
DeepDelta: predicting ADMET improvements of molecular derivatives with deep learning
Many molecular ML models process individual molecules as inputs. This study suggests a model, DeepDelta, that uses a pairwise learning approach: processing two molecules simultaneously and predicting property differences between two molecules. This approach does better than established models (D-MPNN and Random Forest) on many benchmark tasks and outperforms others in molecular property prediction.
ML for Atomistic Simulations
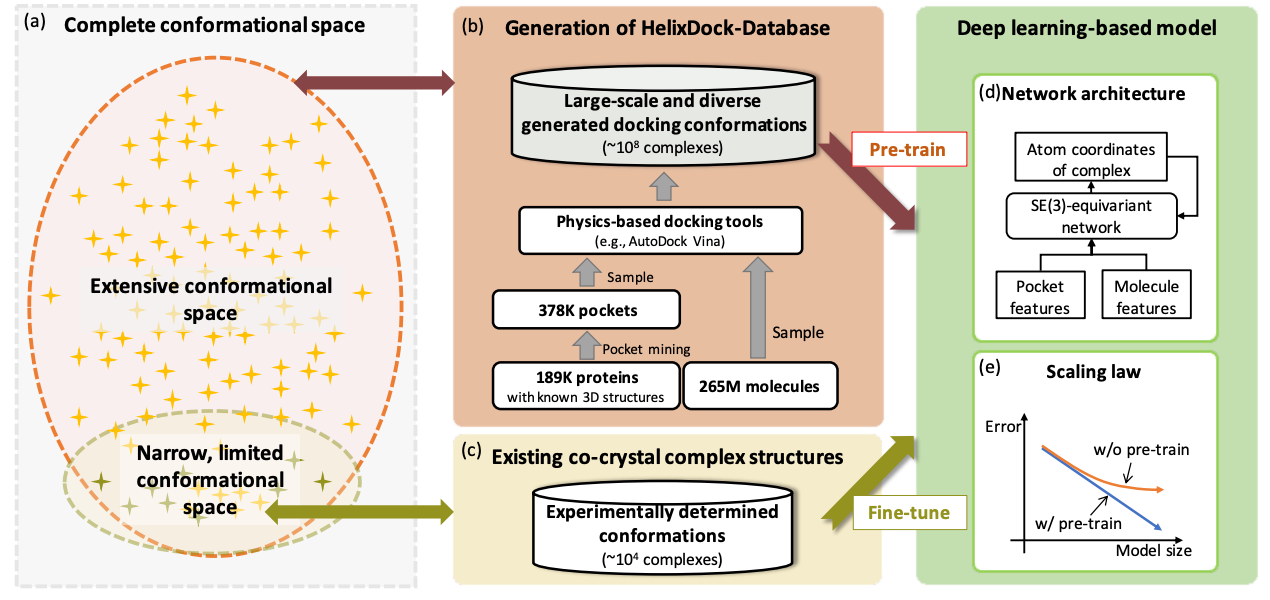
Pre-Training on Large-Scale Generated Docking Conformations with HelixDock to Unlock the Potential of Protein-ligand Structure Prediction Models
Protein-ligand structure prediction is vital in drug discovery, yet conventional tools struggle with accuracy because of limited conformational sampling and imprecise scoring functions. Using deep learning improves predictions, but there may be generalizability issues because of limited validation data. This study proposes HelixDock, which pre-trains on physics-based tools' data and fine-tunes with experimentally validated complexes. It outperforms competitors by over 40% for RMSD.
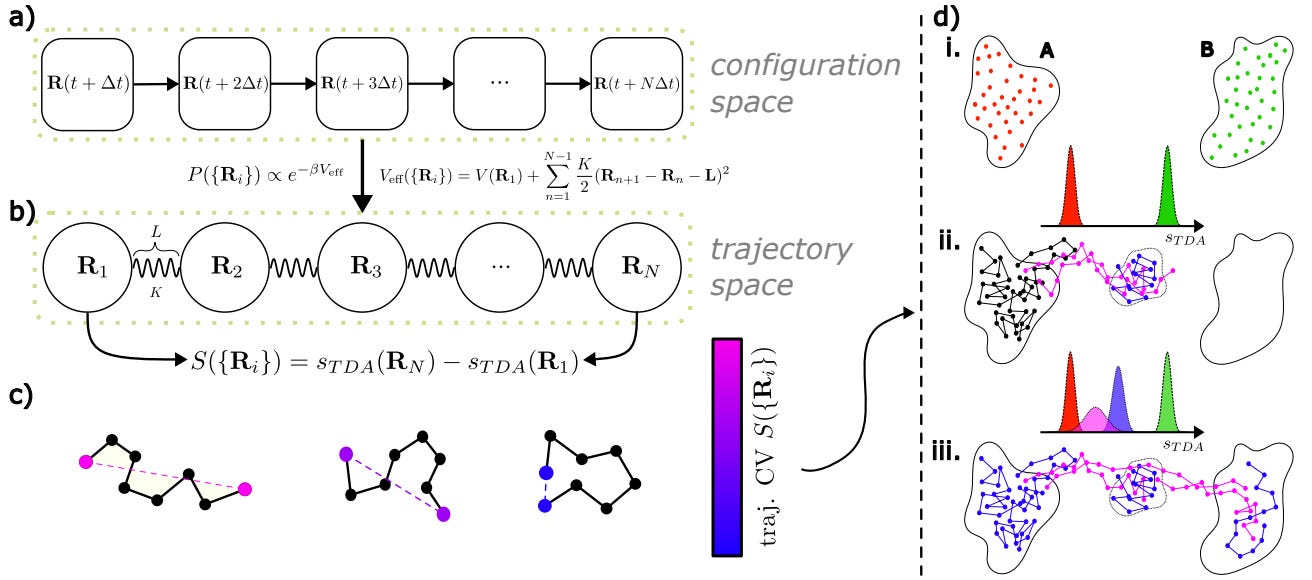
Effective Data-Driven Collective Variables for Free Energy Calculations from Metadynamics of Paths
Various sampling methods predict molecular free energy landscapes using selected collective variables (CVs). The accuracy depends on relevant CVs capturing the system's slow degrees of freedom. ML methods could help, but require high-quality datasets. The study introduces a method to generate such datasets through enhanced sampling simulations, providing an efficient way to predict free energy landscapes.
ML for Proteins
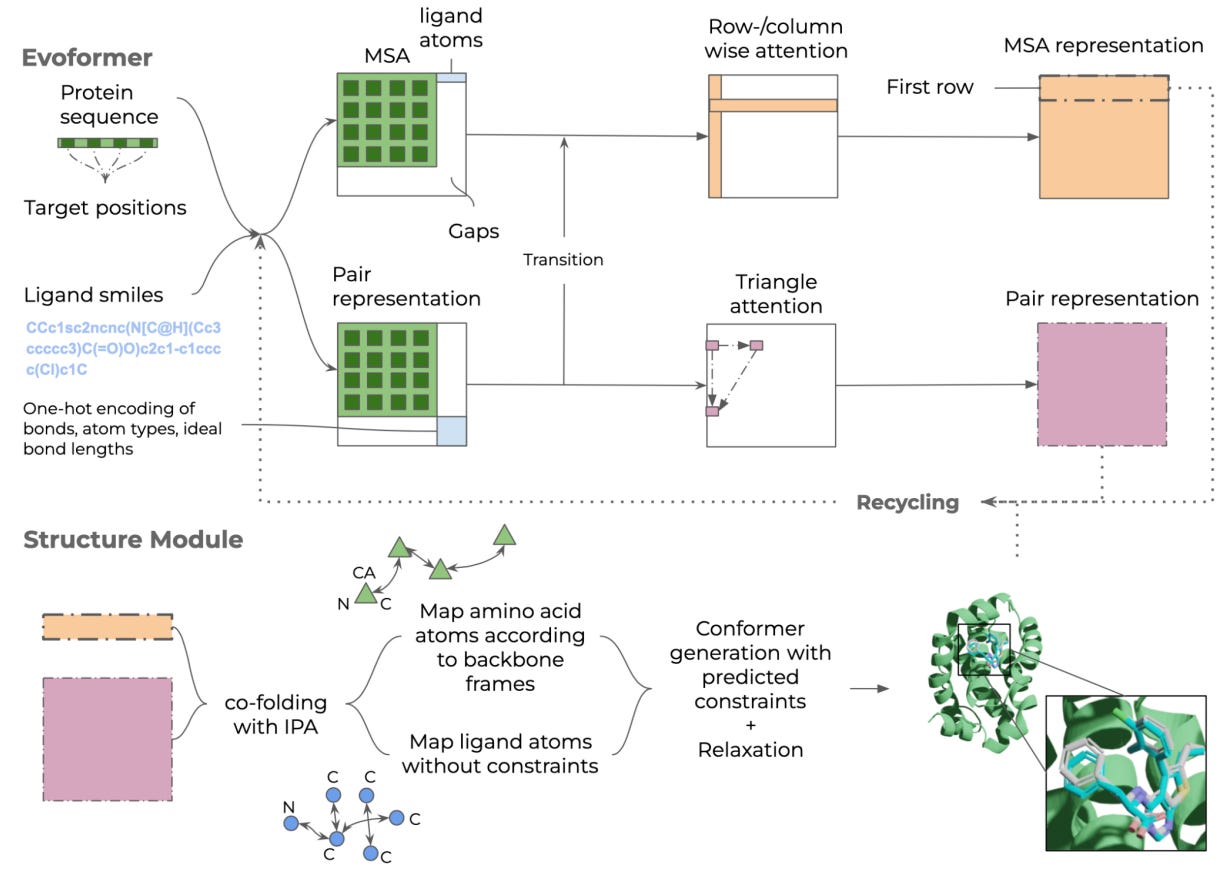
Structure prediction of protein-ligand complexes from sequence information with Umol
Protein-ligand docking is used in drug discovery to select options for experimental testing, but the method needs high-quality protein structure and the protein is treated as rigid. This study provides a model, Umol, that can predict a flexible all-atom structure and seems to predict protein-ligand complexes more accurately than RoseTTAFold-AA and potentially even classical docking methods.
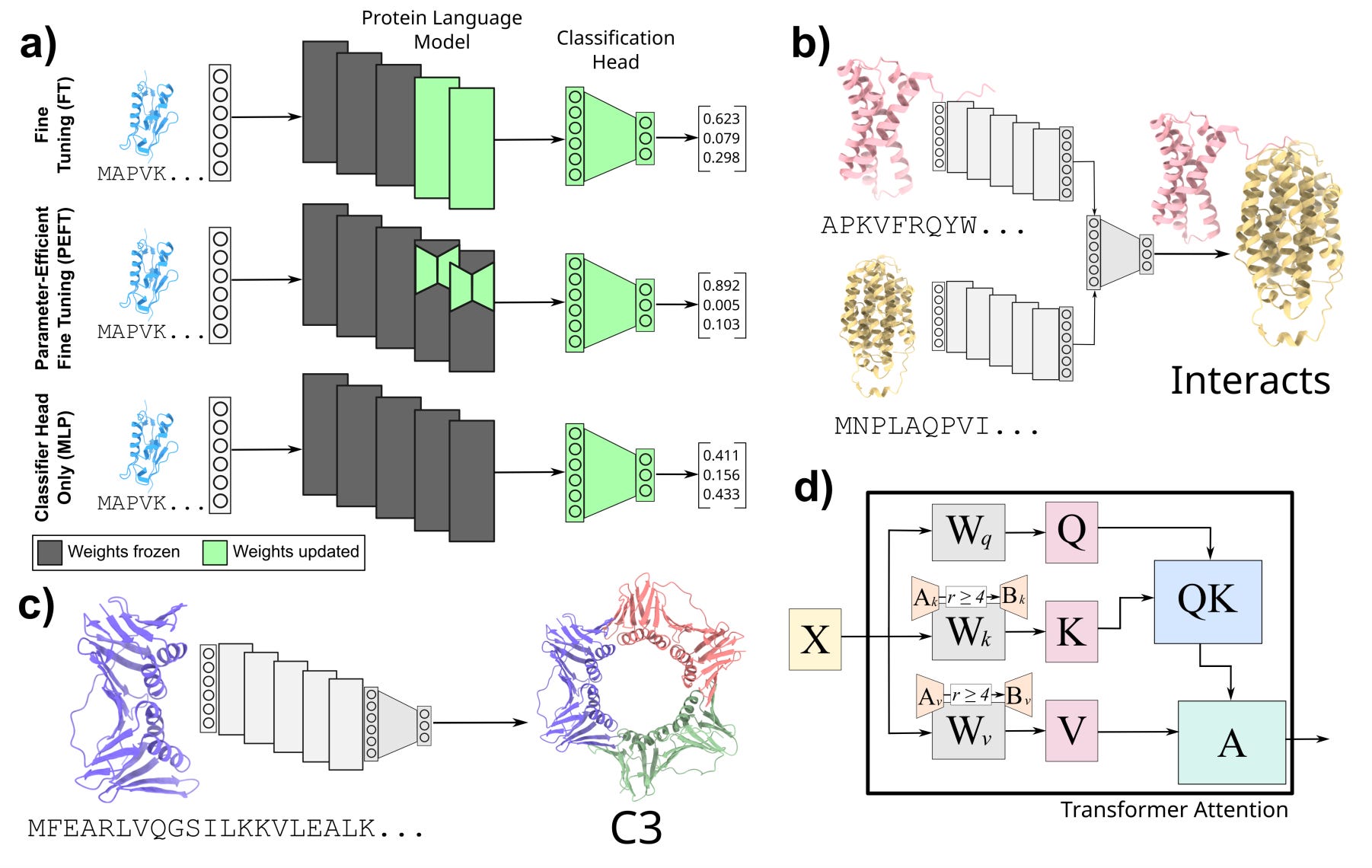
Democratizing Protein Language Models with Parameter-Efficient Fine-Tuning
ML models, especially language models, are getting bigger. As their size increases, the computational and memory footprint of fine-tuning with these models could become a barrier for many research groups. Natural language processing has addressed these issues by parameter-efficient fine-tuning (PEFT) methods. This study applies these methods to proteomics, showing that even with using five orders of magnitude fewer parameters they can still do very well on protein-protein interactions (PPI) prediction.
ML for Omics
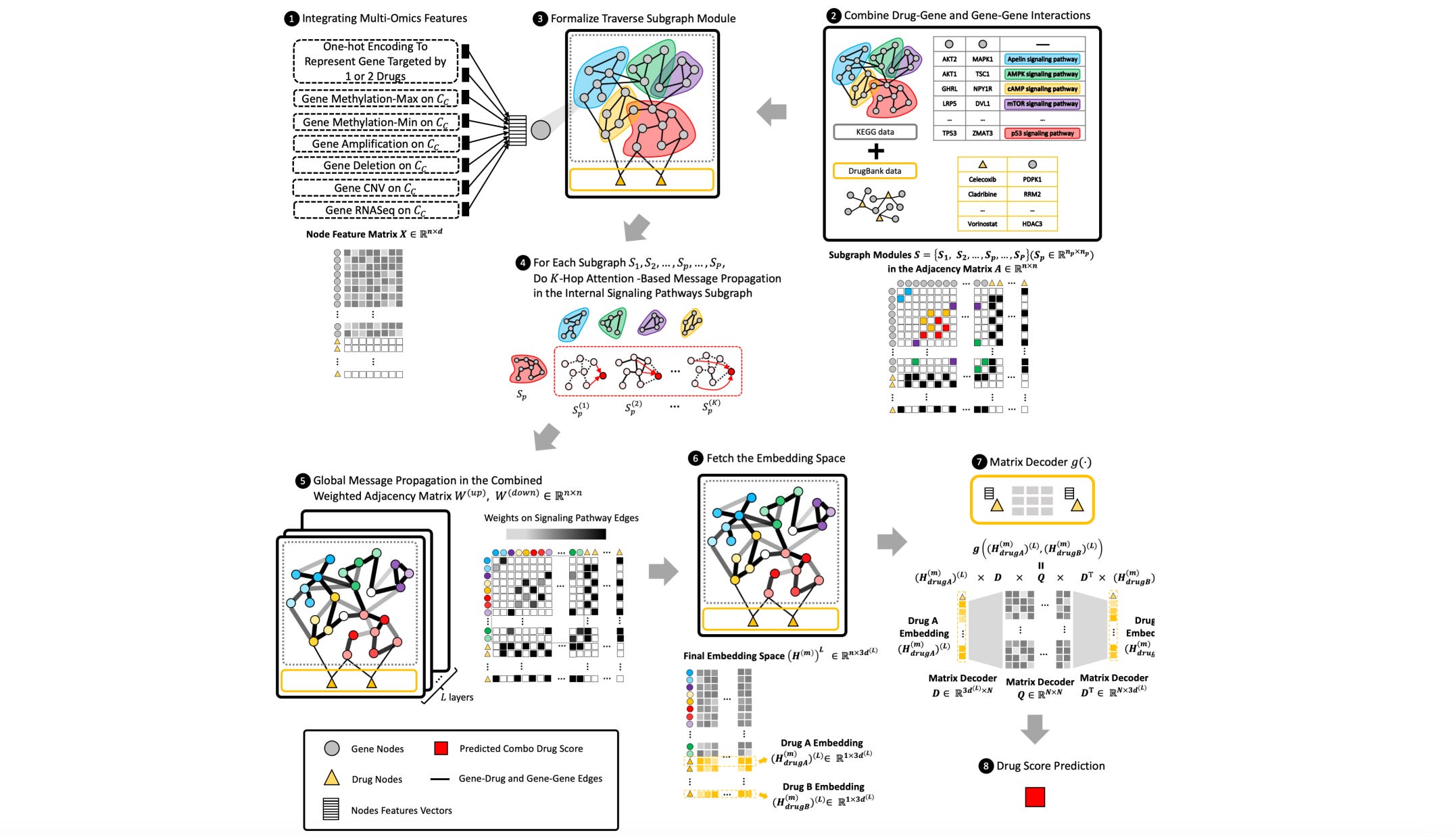
M3NetFlow: a novel multi-scale multi-hop multi-omics graph AI model for omics data integration and interpretation
Multi-omics holds a lot of promise for decoding cell signaling pathways and understanding molecular targets. This study suggests a model to integrate and interpret multi-omics data that does better than existing graph neural network models, and develops a visualization tool to get a better sense of targets and signaling pathways of drugs.
Open Source
CZ CELLxGENE Discover: A single-cell data platform for scalable exploration, analysis and modeling of aggregated data
Biological data standardization is a major issue. Many datasets available via unique portal platforms lack interoperability. This single-cell data resource hosts a curated, standardized, community-contributed dataset that is associated with consistent cell-level metadata.
Random Thoughts
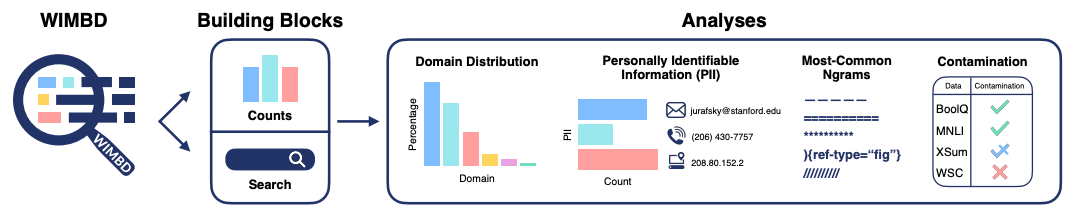
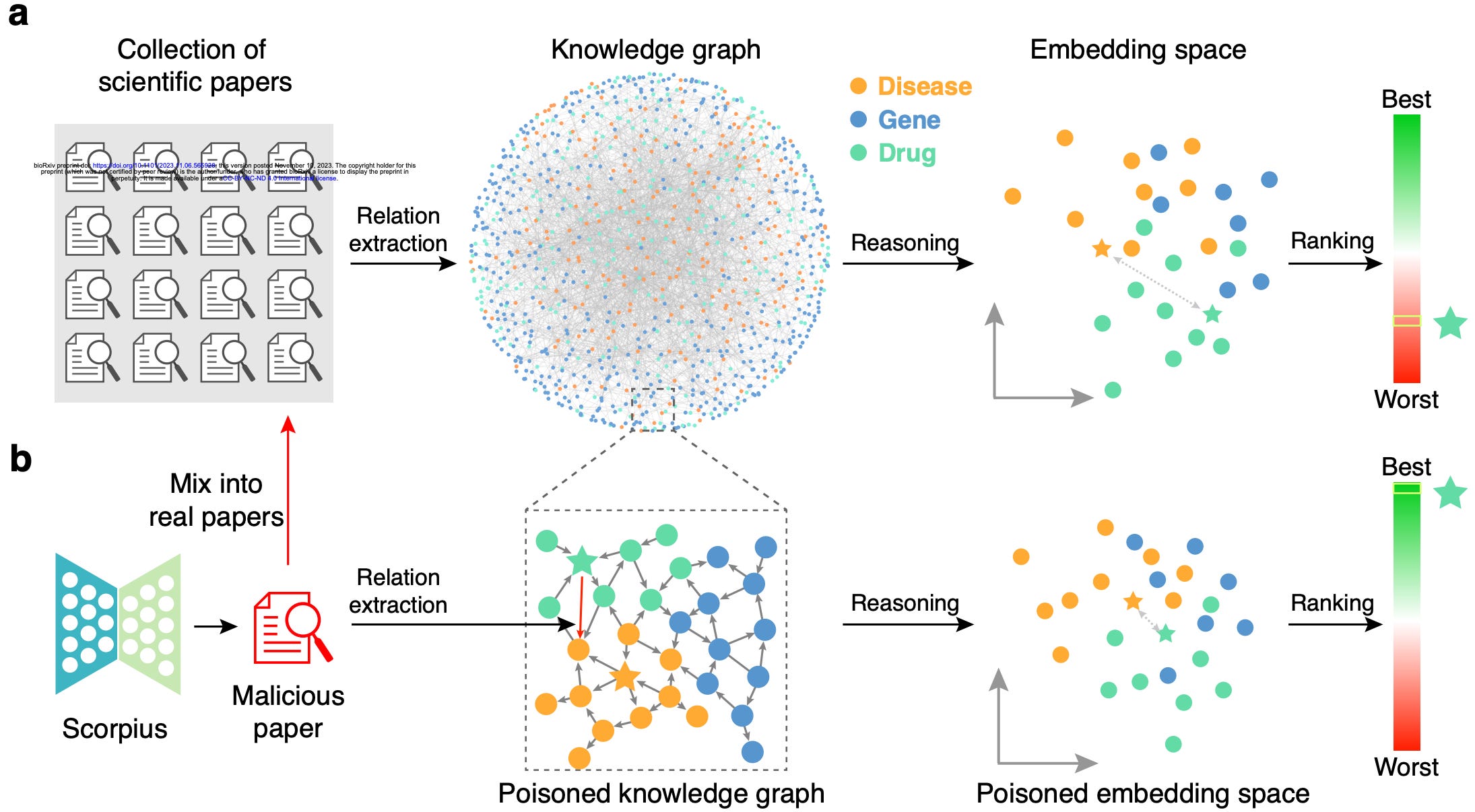
With the mind-boggling sizes of the text corpora used for training large language models these days, it’s hard to know what exactly is in them. What's In My Big Data? presents a platform and analyses to look at and compare the contents of large text corpora. They find a high amount of duplicate, synthetic, and low-quality content, personally identifiable information, toxic language, and benchmark contamination. What sort of effect could this have? Well, if you’re in science it could potentially poison biomedical knowledge graphs, as studied in Poisoning scientific knowledge using large language models. They use a conditional text generation model to make a malicious paper abstract, and find that even one malicious abstract can potentially significantly affect a knowledge graph without being detectable by humans. These kind of studies are important, since we want to ensure that the science we’re doing is accountable and trustworthy.
What's In My Big Data?
Poisoning scientific knowledge using large language models
Think we missed something? Join our community to discuss these topics further!
🎬 Latest Recordings
M2D2
Learning Representations on Biological Data with Weakly Supervised Learning by Ian Smith
LoGG
Bayesian Flow Networks by Alex Graves
Valence Portal is the home of the AI for drug discovery community. Join here and stay up to date on the latest research, expand your network, and ask questions. In Portal, you can access everything that the community has to offer in a single location:
M2D2 - a weekly reading group to discuss the latest research in AI for drug discovery
LoGG - a weekly reading group to discuss the latest research in graph learning
Blogs - tutorials and blog posts written by and for the community
Discussions - jump into a topic of interest and share new ideas and perspectives
See you at the next issue! 👋